MLD
Better understanding of Metachromatic Leukodystrophy
Metachromatic Leukodystrophy (MLD) is a member of the lysosomal leukodystrophy family. It’s a rare autosomal recessive genetic disorder. Its frequency is 1 per 45,000 births. It can begin in childhood, adolescence or adulthood and leads to severe neurological dysfunction affecting motor and cognitive functions that can lead to death.
The gene whose mutation is responsible for Metachromatic Leukodystrophy is the ARSA gene which is located on chromosome 22 (at 22q13.31). This gene codes for arylsulfatase A, an enzyme located in the lysosome of cells. This enzyme causes damage to sulfatides, an important lipid component of the myelin of the brain and peripheral nerves, but also of the neurons of the brain. To date, more than 160 mutations in the ARSA gene have been identified (167 on HGMD).
Alternatively, a very small number of patients carry a mutation in the PSAP gene for Saposin B, a co-activator of the ARSA enzyme.
Mutations
Patients with Metachromatic Leukodystrophy are classified into two main groups according to the mutation they carry: The “O” alleles are associated with extremely low enzyme activity while the “R” alleles are associated with residual enzyme activity.
Carriers of one “O” and one “R” allele account for 50% of Caucasian patients. Other mutations are unique or much rarer.
There is some correlation between patient genotype and the onset of symptoms.
- When patients have both an “O” and an “R” allele, they are most often in the juvenile group.
- Homozygous patients for the “O” allele (who have two copies of the “O” type), with a very low level of ARSA enzyme activity, are found most often in the group of patients with the late infantile form.
- Those with two “R” alleles are found in the adult form group.
So the longer the enzyme remains “active”, the later the disease develops.
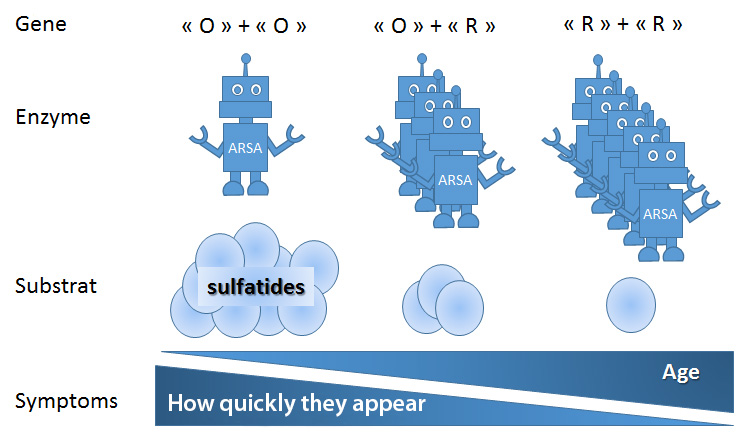
Metachromatic Leukodystrophy: from gene to symptoms. Depending on the genes (“O” and “R” alleles), the ARSA enzyme is more or less active. The less active the enzyme is (“O” + “O”), the more sulfatides that accumulate. The less active the enzyme is, the younger and faster the symptoms appear.
Symptoms of the disease
Metachromatic Leukodystrophy is characterised by progressive motor and cognitive deficits.
Late infantile and early juvenile forms
Late infantile and early juvenile metachromatic leukodystrophies appear before the age of 6 years old. These are the most frequent (60%). They are characterised by motor and cognitive deficits that worsen very rapidly and inexorably. The prognosis is very severe and treatment must be considered very quickly to improve the child’s quality of life.
Late juvenile form
In the late juvenile forms, which appear on average between 6 and 16 years of age, cognitive difficulties may precede motor disorders. The progression of the disease tends to be slower.
Adult form
The adult forms of the disease, declared after puberty, are accompanied by cognitive difficulties and abnormal behaviours that are at the forefront, with isolated peripheral neuropathy in rare cases. In other cases, the symptoms combine motor and cognitive deficits as in the late juvenile form, but with a slower progression.
Diagnosis of the disease
Metachromatic Leukodystrophy is evoked in front of the clinical picture and an evocative aspect on cerebral MRI. It is diagnosed by the detection of a deficiency of the ARSA enzyme in blood cells and abnormal excretion of sulfatides in the urine. These 2 tests are essential for the diagnosis. When the disease is due to a mutation in the PSAP gene, the activity of the ARSA enzyme measured in blood cells or fibroblasts is normal but abnormal excretion of sulfatides is observed in the urine. The diagnosis must be confirmed by sequencing the ARSA or PSAP gene and identifying pathogenic mutations in the gene.
Genetic counselling
When a mutation in both copies of the ARSA or PSAP gene is identified in a patient (an affected patient always has a mutation in both copies of the gene), it is also necessary to look for a mutation in one of the copies of the gene in both parents who are “obligatory heterozygotes”*[1]. In a future pregnancy, there is a 25% risk that the unborn child will be affected. It is possible to offer a reliable prenatal diagnosis of the disease. It is also essential to screen brothers and sisters, especially if they are younger. Indeed, if they have the disease but are still asymptomatic or at the very beginning of their illness with a diagnosis yet to be made, experimental treatment may eventually be offered to them. Each of the siblings of an affected patient has a 25% risk of being affected, a 50% risk of being heterozygous like their parents and a 25% risk of not having any pathogenic mutation in the ARSA or PSAPgene. Other family members (siblings of both parents) may also be heterozygous. Heterozygous people never develop the disease. It is quite common to see subjects with decreased ARSA activity but with no abnormal excretion of sulfatides. These so-called “pseudo-deficient” subjects do not develop any symptoms.
[1] *Compulsory heterozygotes: parents of children with autosomal recessive disease are called “compulsory heterozygotes” because they must carry a mutation.
The simplified mechanism leading to the disease
The ARSA enzyme is at work in the lysosome. It transforms its substrate, 3-O-sulfogalactosylceramide (sulfatide), into another lipid. Sulfatides are important lipid components of myelin, but also of neurons.
The Lysosome: where ARSA works
Lysosome comes from the prefix “lysis” (releasing action) and the Greek suffix “some” meaning “body” ( = soma ). So a lysosome is a eukaryotic cellular organelle[2] which allows the degradation of molecules, mostly lipids. It is a small spherical structure (a vesicle) delimited by a lipid membrane located in the cytoplasm of eukaryotic cells. The membrane contains ion channels (proton pumps and others specific to Cl-chloride ions) that allow the active entry of H+ ions, in order to maintain an acidic pH (between pH 3.5 and 5) within the lysosomal vesicle.
The lysosome has a waste destruction function, where molecules are eliminated by digestion, thanks to enzymes called hydrolases, which are active at acidic pH.
The majority of lysosomal diseases are severe, disabling and degenerative, often leading to premature death.
[2] *Eukaryote: A set of organisms (single or multi-cellular) having a nucleus.
In Metachromatic Leukodystrophy, sulfatides, which are not destroyed by ARSA, accumulate in excess in the white matter cells (oligodendrocytes, microglia) of the brain, brain neurons and Schwann cells that make the myelin of peripheral nerves. This accumulation is toxic to cells, leads to cell death and is responsible for demyelination and destruction of neurons.
Depending on the mutation, the enzyme will be made a little or not at all and therefore be less active (residual) or practically inactive (“O”).
Treatments
- Allogeneic bone marrow transplantation : A hematopoietic stem cell transplant from the bone marrow of a matching donor may be offered to patients with late juvenile or adult forms of the disease. Alternatively, cells from cord blood can be used as a source of stem cells for transplantation. The transplant requires 12 to 24 months to be effective, which does not allow treatment of patients with the late infantile and early juvenile forms, whose development is very rapid. The results are encouraging in the medium term in patients whose disease progresses slowly. Long-term effectiveness is less certain.
- Gene therapy : A clinical study is underway to evaluate the safety and efficacy of gene therapy in children with MLD at different stages, late infantile pre-symptomatic or early juvenile pre-symptomatic, or even early juvenile at the onset of their disease. This trial involves taking hematopoietic stem cells from patients, injecting the normal enzyme gene into these cells in the laboratory with a lentiviral gene therapy vector, and re-infusing the cells (after myeloablative conditioning as in an allogeneic bone marrow transplant). It is ex vivo gene therapy. A first evaluation was carried out in Milan on nine children who had been undergoing gene therapy for a minimum of 18 months (Sessa et al., 2016). They are all alive and the activity of the ARSA enzyme was gradually restored in the circulating hematopoietic cells and in the cerebrospinal fluid. This study, although early, is very encouraging on the safety and efficacy of gene therapy for children treated at an asymptomatic stage (before the appearance of any abnormal neurological signs). Evaluation of the effectiveness of this approach on early juvenile symptomatic forms is still ongoing. However, other questions are emerging: the use of a lentiviral vector allows the insertion of the drug gene into the genome and therefore runs the risk of disrupting the proper functioning of the genome. The long-term risks, including cancerous mutagenesis that may result in the development of Leukemia of this form of ex vivo gene therapy are unknown.
At the same time, a clinical study is being carried out at the Bicêtre hospital and consists of injecting the normal gene for the enzyme (inserted in a viral vector) directly into the brain of the patients (NCT01801709). It is in vivo gene therapy. With this method, a sample is not required and the drug gene enters the patient’s brain cells. If it proves effective, this technique would make it possible to circumvent the risks associated with ex vivo gene therapy. Indeed, the viral vector used allows the therapeutic gene to be inserted into the brain cells but not into the genome of the cells. This approach also has its own risks: the risk of haematoma at the intracerebral injection sites of the vector.
- Enzyme replacement : An alternative approach is to inject the enzyme protein directly into the cerebrospinal fluid from where it could integrate the brain cells and join the lysosomes of these cells to become active. In this therapeutic approach, it is not the gene but the enzyme that is injected, which allows a rapid but temporary action. Indeed, the protein is eliminated quickly and lost over time, hence the need for repeated injections. A clinical study is underway to evaluate the safety and efficacy of this procedure (NCT01510028). The injections are given every two weeks for 38 weeks using a catheter implanted in the lumbar region of the subdural space around the spinal cord where the cerebrospinal fluid is located. The first results are not expected before 2018.
Day-to-day management of the disease
Currently, the treatment of MLD is a supportive symptomatic treatment. It is always possible to offer a comfort solution and all those involved in the care, the medical profession and family members, must work together to identify obstacles and propose solutions.
Metachromatic Leukodystrophy remains an extremely serious disease, except in rare cases where an early marrow transplant can be performed in the late juvenile or adult forms. Gene therapy may offer a long-term alternative for patients with Metachromatic Leukodystrophy who cannot receive a bone marrow transplant. In this battle against time, a combination of effective short-term (enzyme replacement) and long-term (gene therapy) treatments could be considered to treat even the youngest children developing the disease. In all cases, however, these treatments only have a chance of being effective in the early stages of the disease or better when patients are still asymptomatic.