Alexander disease
Alexander disease is a rare genetic disorder in the family of demyelinating leukodystrophies. Its prevalence across the world is not exactly known, but has been estimated at 1 in 2,700,000 in Japan. This neurological disease is most often diagnosed in early childhood, but juvenile and adult forms also exist. Symptoms include mental retardation, epilepsy, and muscle stiffness, and their progression leads to the patient’s death. It is characterised by a degeneration of the white matter and the formation of Rosenthal fibres.
1. Genetic mutation
Alexander disease is caused by dominant mutations in the GFAP gene, which is located on chromosome 17 (at 17q21). This gene enables the production of glial fibrillary acid protein (GFAP). These fibrillary proteins assemble to form intermediate filaments in the astrocytes of the central nervous system.
What is an intermediate filament in plain English, and what is its role in astrocytes.
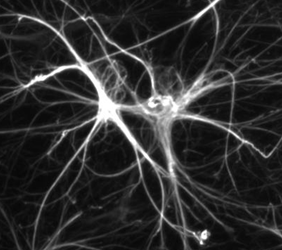
Astrocytes are star-shaped brain cells (hence their name: “astro” for star and “cyte” for cell). They look like stars with beams. These beams are like the arms of the cell. These extensions are necessary for the function of the cell, which has to exchange information with many other cells in its environment: the white matter.
The cytoskeleton [1] is the skeleton of the cell; it is what gives the cell its specific shape.

The intermediate filaments are components of the cytoskeleton. They maintain the cell’s architecture. Type III intermediate filaments are composed of GFAP in astrocytes and other glial cells. These nanofilaments assemble spontaneously and do not require a chaperone protein or energy source. Like a lego model.

GFAP mutations are dominant, meaning that only one mutated copy of the gene causes the disease. In the vast majority of cases, the mutation is sporadic, meaning that it is not inherited from the parents, and it occurs for unknown reasons. Already more than 100 pathogenic variants have been identified.
GFAP mutations, which are found in 90% of patients, are missense mutations. Missense mutations are point mutations in which a letter of the gene is changed (a nucleotide), altering the current assembly of the chain that forms the protein. The result is a modified protein, and in Alexander disease, this protein gains activity. We are talking about gain of function.
2. The simplified mechanism leading to the disease
It is now clear that Alexander disease is an astrocyte disease. In the astrocytes of patients with Alexander disease, the presence of mutated GFAP, and excessive accumulation of GFAP (mutated and non-mutated), are linked to the activation of several cellular stress pathways. Astrocytic hyperactivation could be toxic and the main contributor to pathogenesis. Astrocytes are essential for brain development and function, but until recently these neural cells were largely ignored. The crucial role of astrocytes in the brain is now beginning to be recognised.
Rosenthal fibres
Rosenthal fibres are composed of agglomerated intermediate filament proteins, mainly GFAP. They are formed in astrocytic processes (the arms of astrocytes), and appear as deposits in the cell. These aggregates are sometimes found in other types of disease, but not with the abundance, or particular distribution in the brain, that is seen in Alexander disease. Rosenthal fibre inclusions are visible on brain staining. They look like thick, elongated, worm-like or corkscrew-like bundles, measuring about 10 to 40 micrometres in diameter by 100 micrometres in length.
It is currently not clear what triggers the formation of Rosenthal fibres, and it is not established whether these fibres are protective or toxic. Further research is needed to understand the specific role of GFAP and the mechanisms responsible for the disease.
3. Clinical expression
Alexander disease was first identified in 1949 by Dr Stewart Alexander, an Australian pathologist, who first described an infant case with Rosenthal fibres and loss of myelin. It is a neurodegenerative leukodystrophy, which means that in parallel with the loss of white matter, neurons continually lose their structure and functionality over time.
The symptoms of Alexander disease can vary. They depend largely on the age of onset of the disease but can include: spasms, learning disabilities, feeding problems, increased head and brain size, hydrocephalus (fluid in the brain), delayed development and growth, seizures, reduced mobility, speech problems, mental regression, difficulty swallowing, inability to cough, sleep disorders.
Alexander disease in small children
In 80% of cases, Alexander’s disease manifests itself before the age of 3 years. Young patients develop an enlarged brain and head (i.e. an increase in volume), convulsions, stiffness in the arms and/or legs, intellectual disability and developmental delay.
In rare cases, a neonatal form of Alexander disease occurs in the first month of life and is associated with severe intellectual impairment and developmental delay, accumulation of cerebrospinal fluid in the brain (hydrocephalus) and seizures.
Alexander disease in older children
Fourteen percent of patients develop the first symptoms between the ages of 3 and 12. For these juvenile forms, the symptoms generally include poor coordination (ataxia), difficulty swallowing, speech problems, seizures, etc.
The adult form of Alexander disease
In patients with the adult form of Alexander disease (6% of cases), neurological symptoms and prognosis are variable. Symptoms are non-specific and include, as with the juvenile forms, poor coordination (ataxia), difficulty swallowing, speech abnormalities, and convulsions.
Generally, Alexander disease is less severe when it develops in adulthood. Head size and mental capacity may be completely normal at this stage, but slow mental decline is possible.
Alexander disease in the elderly (65 years and older)
It is extremely rare for Alexander disease to develop this late. If this is the case, the symptoms are often confused with those of multiple sclerosis or a brain tumour. The severity of the disease is often so moderate that Alexander disease is diagnosed after death, when an autopsy reveals unusual protein deposits in the brain.
Symptomatic classification
More recent classification systems have been proposed that rely more on the location of lesions in the brain and spinal cord and, therefore, on the types of symptoms.
Type I patients have a frontal predominance of lesions, all are early and have a more aggressive disease progression. Typically, Type I patients have various developmental delays, affecting both cognitive and motor skills (such as language or walking), followed by loss of bearings, abnormal increase in head size and often convulsions.
Type II patients have a predominance of the hind brain, with lifelong appearances and slower disease progression. Type II patients have more difficulty walking. Many patients have problems with excessive vomiting, difficulty swallowing and speaking
Type I patients predominate in the literature, but this probably reflects a verification bias, as adult patients in particular are often misdiagnosed with other conditions, such as Parkinson’s disease or multiple sclerosis.
4. Diagnosis of the disease
Although there are symptoms and clinical elements that may point to Alexander disease, the diagnosis can only be validated by genetic testing. People with symptoms are referred to a specialist in genetics and metabolism who can ensure that a correct diagnosis is made.
5. Genetic counselling
To date, more than 100 different genetic mutations have been identified. In most cases, the genetic mutation causing the disorder is spontaneous, meaning that it is not a mutation passed on by either parent. Therefore, it is unlikely that other children of the same parents will have Alexander disease. Siblings of children with Alexander disease are unlikely to have the disease themselves. Only a few family cases have been reported. It is important to talk to a geneticist or expert on Alexander disease to discuss the likelihood that the disease will be genetically transmitted to other family members.
6. Treatments
To date, there is no definitive treatment for Alexander disease. However, regardless of the form diagnosed, it is important that comprehensive medical care be put in place quickly. Through proactive and comprehensive medical care, patients can avoid unnecessary suffering and complications and have the best possible quality of life.
Particular attention must be paid to the following:
General care
Ergotherapy and physiotherapy
Nutrition
Speech therapy
Antibiotics for any developed infection
Anti-epileptic drugs to get seizures under control
Research
With the absence of a definitive treatment, research continues. An even better understanding of the disease is needed in order to consider treatments.
• Antisense oligonucleotides
To treat Alexander disease, researchers are investigating the use of antisense oligonucleotides (ASOs), a technology that suppresses the expression of a particular gene. These treatments are being developed for several diseases, and a first ASO has been approved in Europe and the United States for the treatment of spinal muscular atrophy, a rare neuromuscular disorder.
For Alexander disease, one ASO treatment is already being studied in animals, and the initial results are encouraging. A single injection of ASO into the cerebral ventricle works within a few weeks. Rosenthal fibres disappear and several markers return to near normal levels. These results have generated significant interest in the clinical community and could lead to a formal clinical trial, but much work remains to be done, including assessing the ability of ASOs to produce improvements in motor and behavioural phenotypes.
• EJP Alexander Disease
As part of its mission to support leukodystrophy research, ELA International is participating in a European Joint Programme for Rare Diseases (EJP Rare Diseases) focusing on Alexander disease. The objective of this 3-year project is to better understand the process of disease initiation and development. It is being led by Professor Elly Hol from the University of Utrecht in the Netherlands and involves 7 research teams from 6 different countries (Sweden, Czech Republic, Israel, Spain, Luxembourg, New Zealand). The synergy of expertise and knowledge and the cross-border enhancement of resources are strengths of these European programmes with which ELA is associated.
Conclusions
There are still many grey areas around Alexander disease, both in terms of understanding the mechanisms of the disease and in the search for treatments. Only 500 cases have been described worldwide, making it an extremely rare disease and complicating research. Today’s physicians are committed to better managing these patients through proactive and comprehensive medical care and to providing the best possible quality of life for these patients. Research continues, with the support of ELA, to hope for solutions tomorrow.
[1] Cytoskeleton: organised set of biological polymers of a cell that give it most of its mechanical properties.