ALD/AMN
Adrenoleukodystrophy
X-linked adrenoleukodystrophy (ALD) is a worldwide genetic disease, rare and serious, of the family of leukodystrophies. Its incidence of 1 for 17 000 births makes it, however, the most common type of leukodystrophy. It is a metabolic disease which affect the brain, the bone marrow and peripheral nerves, but also the adrenal (and the testis) as part of its name « adreno- » refers to.
Adrenoleukodystrophy can be visible during childhood, adolescence or adulthood and can present itself under different forms:
Most commonly the disease is slowly progressive and affects the bone marrow and, to less extend, the peripheral nerves in men and women: this is adrenomyeloneuropathy (AMN) which strike the motor functional prognosis of the lower limbs.
More rarely under the form of a rapidly evolving brain disease (cerebral ALD) in boys and adults men, which ends dramatically in absence of bone marrow transplant.
1. Genetic mutation
X and Y chromosomes
Chromosomes are super molecules present in the nuclei of cells. They encompass the DNA, the genetic heritage of each parson.
In human, each cell contains 46 chromosomes, grouped in 23 pairs. A pair includes a copy from the mother and a copy from the father. A cell has two copies of each gene that do not have to be identical. The X and Y chromosomes are called sex chromosomes. They form the 23rd pair of chromosomes. Their names come from the fact that men and women are not identical on that 23rd pair. Women have two copies of X and the men have one X and a Y. Thus the women will always give an X to her children and a man either an X or a Y, which will determine the sex of the child. Both men and women have an X in common, but the men only have one copy when women have two. And unlike men, because they do not have a Y, women do not have any of the genes from the Y chromosome.
The gene with a mutation in ALD is ABCD1, located on the X chromosome (on Xq28). Men having only one copy of the X chromosome, they only have one copy of the gene ABCD1 and will always suffer from the consequences of the mutation when it exists. Women have two copies of the X chromosome, hence two copies of ABCD1 (one from their mother and one from their father). But only one of the X chromosome and one copy of ABCD1 will be functional in the cells of the different organs in the body, in different proportions. For example in the bone marrow, a woman heterozygote carrier of the mutation, can have 80% of her cells expressing the gene ABCD1 wildtype or inversely 75% of cells expressing the mutated ABCD1 gene. Heterozygous women will always have at least 10 to 20% of their cells expressing the normal gene ABCD1. This explains probably why heterozygous women never develop the cerebral lesions and also why they do not all develop AMN symptoms in adulthood.
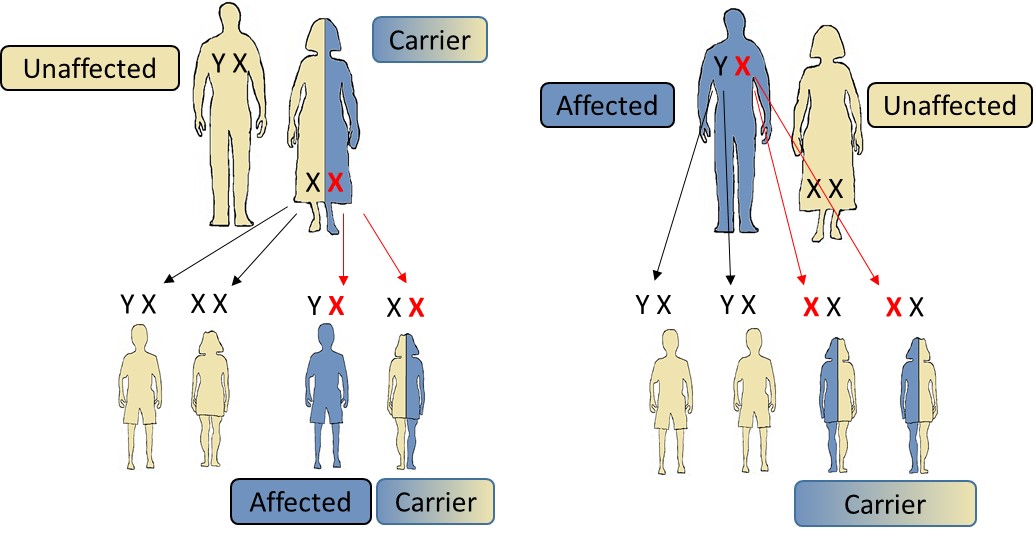
Figure 1: Genetic transmission. If the mother is a carrier (left) of an ABCD1 gene mutation, each daughter will have a risk on two to carry the disease, and a chance on two to be free of the mutation. Each son will have a risk on two to carry the mutation and to develop the disease, and a chance on two to be free of the mutation. If the father is affected (right), all his sons will be exempt of the mutation and all his daughters will be carriers of the mutation responsible for the disease.
2. Simple mechanism leading to the diseas
The peroxisome
Adrenoleukodystrophy is a peroxysomal disease, which means that it results from the impairment of small cell components called peroxisomes. The peroxisomes are responsible for the degradation of the very long chain fatty acids (VLCFA). The gene ABCD1 has been identified in 1993 by the teams of Pr. Aubourg and Pr. Mandel. It codes for the protein ABCD1 (or ALDP), a transporter responsible for the entry of very long chains fatty acids into the peroxisomes, where they can be degraded.
In adrenoleukodystrophy, the ABCD1 gene mutation leads to an inappropriate transport and does not allow the very long chain fatty acids to enter the peroxisome to be destructed by oxidation. This is what leads to their accumulation in every cell of the body and in the blood. We do know today, however, that an ABCD1 mutation can lead to an anomaly of certain cells functions without it being the results of the VLCFA accumulation.
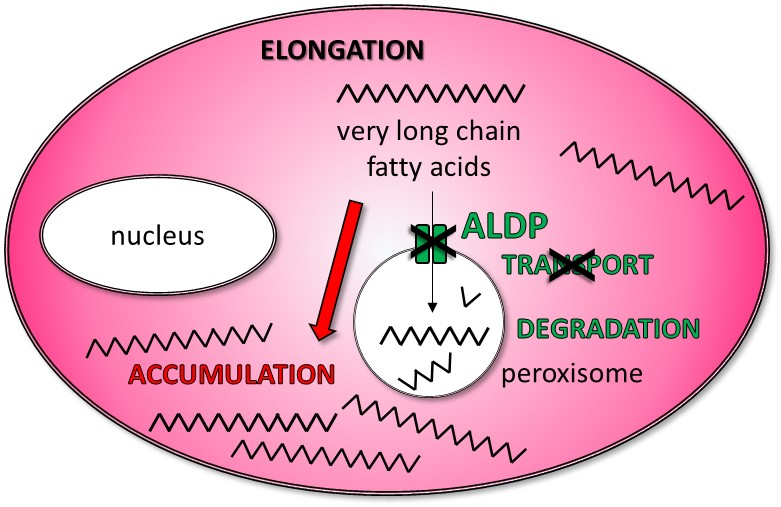
Figure 2: Metabolic anomaly. In cells, peroxisomes are compartments in which the very long chain fatty acids (VLCFA) are degraded. The protein ALDP, made from the gene ABCD1, allow the transport of VLCFA into the peroxisomes. In patients affected by ALD, the ABCD1 gene is mutated and the protein ALDP coded is not functional: the VLCFA cannot enter into the peroxisome to be degraded: they accumulate in the cells, linked to other lipids and proteins.
Accumulation of very long chain fatty acids
In experimental cell culture conditions, very long chain fatty acids seem to be toxic for oligodendrocytes, the myelin forming cells. Their accumulation lead to cell death. It disturbed calcium homeostasis and lead to mitochondrial impairment and production of reactive oxygen species. When oligodendrocytes die, the myelin sheets (which are in reality extensions of oligodendrocyte membranes enrolled around the axons) cannot be maintained, which can lead to the sheets destruction, like what is observed in cerebral ALD. In AMN, VLFCA accumulation in oligodendrocytes of the bone marrow does not lead to oligodendrocytes death but to an impairment of the axons without significant demyelination. When VLCFA accumulate in the cell, long chain fatty acids with shorter chains (18 ; 20 or 22 carbons) are accessible to the molecule responsible to make them longer, increasing even more the quantity of VLCFA. Like a vicious circle.
Breakdown of the blood-brain barrier
It has been demonstrated that the disruption of the blood-brain barrier, a characteristic of adrenoleukodystrophy visible on MRI, occurs in areas of the brain before any demyelination (Musolino et al., 2015). The inflammation which leads to the degradation of the myelin (by infiltration of leukocytes) appears to be a direct consequence of the breakdown of the blood-brain barrier, and is not a consequence of VLCFA accumulation like it was first thought. ABCD1 is very strongly expressed in endothelial cells (and astrocytes) of the blood-brain barrier. It was shown that in ALD patients, in the absence of ABCD1, proteins normally located at tight junctions and sealing the blood-brain barrier, were not there properly located. A passage is thus opened and inflammatory molecules and cells can penetrate into the brain. The absence of a functional ABCD1 protein could be responsible for the opening of the blood-brain barrier. In the brain, the breakdown of the blood-brain barrier could be involved in the development of cerebral form of adrenoleukodystrophy.
3. Clinical expression
At birth, boys and girls do not have symptoms and their apparition is unpredictable. However, all adult males with a mutation of the gene ABCD1, who did not develop the cerebral form during childhood, will develop symptoms of AMN by the age of 55. As described above, heterozygous girl and woman carriers never develop brain damage. However, it is known today that 80% of them eventually develop symptoms of AMN when they reach the age of 60.
There is no correlation between genotype (type of mutation of the gene ABCD1) and onset of symptoms. In other words, within the same family, boys or adult men (brothers, uncle-nephew, for example) can develop the disease differently. From one family to another, even if the mutation of the gene ABCD1 is identical, patients may have different manifestations of the disease. Although they are not known specifically, external factors seem responsible for triggering brain inflammation. Brain trauma or stroke, even minor, can trigger cerebral demyelination in patients presenting only symptoms of AMN or no signs of the disease. Genetic characteristics unique to each patient, and different mutation of the gene ABCD1, can also explain the resistance to develop the most severe form of ALD (cerebral demyelinating form) or the appearance of AMN at a later age or in a less severe form.
Cerebral ALD
All boys with ALD are born without symptoms, and cerebral ALD most commonly appear between 5 and 12 years old, never before 4 years old. Demyelinating lesions first appear on the MRI images and first develop slowly over months or even years, without any recognizable symptoms. Only in a second phase appear simultaneously clinical signs, associated with a significant and rapid increase in cerebral demyelinating lesions. Without bone marrow transplant, and after a major motor and intellectual degradation, the status of children may stabilize for years or even decades. Many, however, especially younger boys, may die from secondary complications, more often respiratory.
The clinical symptoms depend on the location of the demyelinating lesions. In general, the disease progresses more quickly when it starts early. The most common first symptoms are: school problems, attention deficit, difficulties to orient themselves in space, graphics difficulties (drawing, writing), difficulty dressing (dyspraxia), difficulties to understand (these boys often ask twice as if they did not hear), anxiety attacks due to the fact that they see less, understand less and do not understand why.
In adults, at least 65% of men between 20 and 55 years old also developed a cerebral form of ALD visible on brain MRI. In half these cases, the brain damage spontaneously stabilizes with little consequence on motor or intellectual capabilities and patients can live a normal life (unless they also have signs of AMN, which is almost always the case). For the other half, the evolution of the cerebral form is identical to that of the boys, the initial period of slow worsening of intellectual, visual and auditory functions being however longer than for boys. All adult men with demyelinating brain injury also exhibit signs of spinal cord defect.
In some cases, the cerebral form does not progress immediately to an inflammatory stage (less than 5%). In these chronic forms, brain demyelinating lesions evolve very slowly and the patients can be stable for decades. These patients do not have visual or motor impairment except when adrenomyeloneuropathy appears in adulthood. However, they have significant intellectual deficits that complicate their adaptation to a normal school or professional life.
These cerebral forms are not described in women.
Adrenomyeloneuropathy
The first symptoms of adrenomyeloneuropathy usually appear in men between 20 and 30 years of age. Most often, they include difficulties to run, walk long distances or walk in rough terrain. Motor disability progresses slowly over decades. Adrenomyeloneuropathy is characterized by spastic paraparesis (motor deficit of the lower limbs with stiffness) associated with balance disorders (sensory ataxia), and difficulty urinating. An adrenal insufficiency, clinical or only biological, is very common among these men.
Female carriers also develop symptoms of adrenomyeloneuropathy. The age of onset is older, between 40 and 50 years. Initial symptoms are the same as in men, but women are more often affected by balance disorders than by stiff legs, and have more neurogenic pain (like an electric shock, or crushing …) in the legs. Urinary problems are also more frequent and severe and sometimes associated with anal sphincter control problems.
Symptoms of peripheral neuropathy are rare, both in men than in heterozygous females.
Adrenomyeloneuropathy most often develops slowly over decades with apparent stabilization periods that can last for years. The upper limbs are never affected. Mental abilities are not altered
Adrenal insufficiency (Addison’s disease)
About 70% of adrenoleukodystrophy male patients eventually develop adrenal insufficiency at a point in their lives. It can precede the neurologic symptoms of adrenoleukodystrophy (sometimes for decades) or occur simultaneously.
It can be detected from the age of 3-4 years-old, by a brown pigmentation stronger in the face, neck and back of hands, on scars or skin folds of the fingers, by increased fatigue, digestive disorders, nausea or lack of appetite. Adrenal insufficiency should be a meaningful sign of a possible adrenoleukodystrophy.
In female carriers with adrenomyeloneuropathy symptoms, the existence of adrenal insufficiency is very rare.
Testicular failure
The men caring mutation often show biological signs of testicular failure without clinical signs. Men may present erectile dysfunction due to involvement of the spinal cord. Very rarely, they are due to testicular hormonal deficiency that causes a decrease in testosterone and therefore of sex drive. Female carrier show no abnormal ovarian function.
Female carriers:
Historically, adrenoleukodystrophy was considered a disease affecting only boys and men. It has become clear over the past fifteen years that at least 80% of female caring the mutation responsible for the disease eventually develop neurological symptoms of AMN when they reach the age of 60. Their symptoms are usually less severe than in adult men. The percentage of female carriers who develop a symptom, gradually increases with age, reaching 30% of female carriers at the age of 20, and over 80% of women after 60 years old.
4. Diagnosis of the disease
Adrenoleukodystrophy is suggested by neurological or endocrine clinical patterns and white matter abnormality in brain MRI when present. It is diagnosed by measuring the concentration of very long chain fatty acids in the plasma. Three levels are measured: the content of saturated fatty acid in 26 carbons, the ratio of saturated fatty acid levels at 24 and 22 carbons, and the ratio of saturated fatty acid levels at 26 and 22 carbons.
This diagnosis is completely reliable in men. In heterozygous females, the diagnosis is falsely negative in 20% of cases. Genetic analysis (identification of ABCD1 gene mutation) is required to confirm or refute the diagnosis.
The biochemical changes (increase in the plasmatic concentration of very long chain fatty acids) appear before the first symptoms occur, allowing screening for adrenoleukodystrophy in newborn boys and girls.
5. Genetic counseling and screening for the disease in each family
More than 700 different mutations have been identified. With few exceptions, each ALD family has its own ABCD1gene mutation. Screening every family is necessary:
That the disease first affects a boy (cerebral form or adrenal insufficiency), a man or an adult woman (AMN), it is very important to screen all boys and adult men at risk of being affected in each family. This is the only way to diagnose them in time if they are still completely asymptomatic (normal brain MRI) or starting to develop brain lesions without any symptoms. And to offer them the only treatment that can stop the progression of this brain disease: allogeneic bone marrow transplantation or gene therapy.
Screening is also essential to identify all women at risk of caring a mutation (heterozygotes), informing them of the risk to their future children and about prenatal diagnostic modalities.
It is also necessary to identify men with child bearing age daughters who have not yet developed the disease (for men, the first signs of AMN can sometimes appear after the age of 45-50 years old).
This screening allows to identify male ALD patients suffering from adrenal insufficiency, which, not diagnosed and not treated in time, can be very serious and sometimes fatal.
The screening is based on the measure of VLCFA in men and the search for ABCD1 gene mutation in women.
6. Newborn Screening
Adrenoleukodystrophy can also be screened in all newborns, as early as 3 days of life, from a blood sample deposited on a filter (Guthrie sample). Systematic newborn screening programs for ALD are in place in the USA and will soon be established in the Netherlands. Neonatal screening for adrenoleukodystrophy, technically possible today, does not exist in France.
7. Treatments
• Allogenic bone marrow transplant
An allogeneic bone marrow transplant can be proposed for the treatment of early stages of the cerebral form of the disease. It is the only treatment that, when done at the very beginning of the disease (in practice when the patients have no symptoms), stabilizes demyelinating brain lesions. A bone marrow transplant is ineffective, and may even be harmful, at an advanced or late stage of the disease. Grafting is only possible if a compatible donor (bone marrow or cord blood) is found (see the previous issue of ELA info). Bone marrow transplantation has the same efficacy in adult males as in boys. In boys, when the donor is not a healthy brother or sister, the risk of mortality due to graft complications (rejection, graft versus host reaction) is in the range of 12-13%. For men, the risk of mortality due to complications of the graft is much higher: around 35%, in particular because of fatal bacterial complications that are no longer observed in children.
• Gene therapy
Gene therapy is still in trial and consist of an autologous transplantation of bone marrow stem cells from patients and their repair in the laboratory using a viral gene therapy vector. The first trials (4 patients in a first Phase I / II trial and 17 patients in a second Phase II / III trial) indicated a very good gene therapy efficacy comparable to that of bone marrow transplantation, without any of the complications. If the results of the Phase I / II trial are confirmed, gene therapy could be proposed, relatively quickly, to boys at an early stage of their cerebral disease for whom no compatible donor (healthy brother or sister) is available. Gene therapy will also be offered in a second stage to adult men with potentially severe progressive cerebral form.
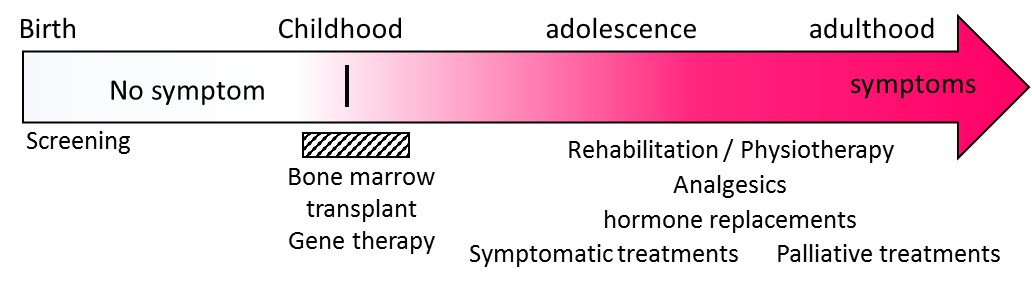
Figure 3: Evolution and treatments. At birth, people with ALD have no clinical symptoms. Very long chain fatty acids can be detected at 3 days of life. With the apparition of the very first symptoms of the cerebral form of the disease, a bone marrow transplant can be proposed. If there is no matching donor available (brother or sister not attained), gene therapy may be considered. Symptomatic care and hormone replacement therapy are possible at any time. Psychological care and palliative treatments can be offered.
• Treatment of adrenomyeloneuropathy
Treatment of adrenomyeloneuropathy is based on symptomatic management of patients: active and passive motor rehabilitation, treatment of urinary disorders, spasticity, lower limb pain, erectile dysfunction. Maintaining regular bi-weekly walking activity remains the most effective “treatment” for AMN. By itself, it significantly improves the motor deficit of the lower limbs
• Treatment of adrenal insufficiency
Treatment of adrenal insufficiency is possible via hormone replacement therapy with adrenal corticosteroids (prescription of hydrocortisone or even fludrocortisone). This extremely simple, oral and daily treatment is essential and should never be interrupted.
Lorenzo’s Oil
This diet is based on a dietary restriction of very long chain fatty acids and the intake of oil enriched in oleic acid and erucic acid (“Lorenzo oil”). It makes it possible to normalize plasma levels of very long chain fatty acids in three months, but it has no effect in the brain forms of ALD and there is no scientific evidence of a modifying effect of Lorenzo oil in AMN or even a real preventive effect. Finally, it must be made clear that Lorenzo oil is indicated only in the treatment of the biochemical defect of ALD and plays no role in other demyelinating or progressive neurological pathologies in which people can metabolize very long chain fatty acids.
• Treatment of Advanced Cerebral Forms
Palliative treatments may be proposed. They are critically important in improving the quality of life of affected children and adults: it can help with pain, spasticity, be the treatment of orthopedic complications, allow adequate nutritional intake using a gastric tube.
Despite numerous attempts for over 30 years, no treatment has been shown to be able to stabilize an advanced cerebral form of ALD.
Prenatal diagnosis
A prenatal diagnosis can be proposed to all female carrier to determine whether the unborn child is a boy, and if he is, determine if he is carrying the mutation of the ABCD1 gene and therefore at risk of developing during childhood or in adulthood one of the neurological forms of the disease.
This screening requires a prerequisite: that it has been demonstrated that the female carrier is indeed carrying a mutation of the ABCD1 gene. This test simply requires a blood test.
The procedures for prenatal diagnosis are as follows:
- if the period is late, confirm by a blood test that the woman is well pregnant and confirm that she is a mutation carrier, do a quick ultrasound in order to date exactly the beginning of the pregnancy;
- explain to the expectant mother, to the couple, the risks of having a carrying daughter, to have an affected child and, if so, what can be offered today as treatment;
- around 7-9 weeks of pregnancy, determine by a simple blood test if the future mother is expecting a boy or not. The test relies on the identification of Y chromosome sequences in the maternal blood (there always are fetal cells that pass through to the blood of pregnant women). This test is totally reliable;
- between 11 and 13 weeks of pregnancy, a trophoblast puncture is performed. This procedures consist to take a tiny bit of the placenta that has the same genetic origin as the fetus. This puncture involves a minimal risk of spontaneous abortion;
- then the mutation of the ABCD1 gene is investigated on this trophoblast sampling. The result is usually given 8-10 days after;
- much more rarely, in practice when the diagnosis of pregnancy was made too late to propose a trophoblast puncture in time, an amniotic puncture takes place between 15 and 18 weeks of pregnancy. This provides cells of the fetus on which are made: a) a karyotype (examination of the chromosomes) in order to determine that it is a boy; B) and if so, a search for the mutation of the ABCD1.
If the unborn child is carrying the mutation of the ABCD1 gene, the woman, the couple, may be offered a voluntary termination of pregnancy.
The preimplantation diagnosis (implantation of an egg obtained by in vitro fertilization after having verified that this egg composed of a few cells does not show a mutation of the ABCD1 gene) is in practice now reserved for women who have already done at least one, or even two voluntary interruptions of pregnancy following a prenatal diagnosis. The reason is simple: there are very few centers competent to make this diagnosis in France, this complicated procedure is not without risk and the ALD is only a rare disease among many others. There are many other requests from other couples concerned with other rare diseases.
• Psychological support
Psychological care must be provided to accompany not only the patients but also the siblings of the patients, their parents, the spouse and often several members of the same family.
• Managing the disease on a daily basis
Physiotherapy support as well as the management of urological complications can be proposed for the management of disorders related to adrenomyeloneuropathy, in order to maintain a quality of personal and professional life.
Conclusions
Many grey areas still exist for ALD in terms of the understanding of certain mechanisms (why 2/3 of the male patients develop brain damage in childhood or adulthood and other do not …) and for the treatment of advanced cerebral forms.
Better knowledge of the disease has allowed the development of a test that could identify all newborn boys, allowing to follow them very early and to offer them a treatment that we know today is only effective when it is proposed early. Newborn screening for ALD is not carried out in France and will be a new fight for ELA. It would eradicate advanced cerebral forms for which we still cannot do anything.