PMD et autres leucodystrophies hypomyélinisantes
Connaître la maladie de Pelizaeus-Merzbacher (ou PMD)
Article revu par le Pr. Nicole Wolf
La maladie de Pelizaeus-Merzbacher (ou PMD) est une maladie génétique rare de la famille des leucodystrophies. Sa fréquence est de 1 pour 100 000 naissances. Elle fait partie des leucodystrophies hypomyélinisantes, ces maladies de la substance blanche caractérisées par un déficit permanent de myéline au niveau du cerveau.
La maladie de Pelizaeus-Merzbacher tient son nom de deux médecins allemands l’ayant décrite au début du vingtième siècle. En 1885 déjà, une famille avait cinq garçons présentant un mouvement oculaire oscillant involontaire, une spasticité dans les membres, un contrôle très limité de la tête et du tronc et un retard dans leur développement cognitif. Vingt-cinq ans plus tard, en 1910, le réexamen de la famille a montré que 14 membres de cette famille étaient atteints de la maladie, dont deux filles, et que tous descendaient d’une même parente. Il a aussi été noté à cette époque que la maladie n’était jamais transmise d’un père vers ses fils, ce qui est connu depuis comme une caractéristique des maladies génétiques dont le gène responsable est porté par le chromosome X.
La maladie de Pelizaeus-Merzbacher se présente sous différentes formes en fonction de l’âge d’apparition des premiers symptômes : une forme néonatale, et une forme dite « classique » qui survient avant l’âge d’un an. Deux autres formes moins sévères ont été décrites : la paraplégie spastique de type 2 (qui inclue la forme récemment décrite, HEMS pour – Hypomyelination of Early Myelinating Structures en anglais) et le phénotype PLP1 nul.
Le gène dont la mutation est responsable de la maladie de Pelizaeus-Merzbacher est le gène PLP1 qui est effectivement situé sur le chromosome sexuel X (en Xq22.2). Pour cette raison, les hommes et les femmes ne déclarent pas la maladie de la même façon, et la maladie affecte typiquement des garçons ou des hommes. Ce gène code pour la protéine protéolipide 1 (PLP1) : 188 mutations entrainant la maladie ont été décrites à ce jour.
« PMD-like »
Un faible pourcentage de patients présentant un phénotype caractéristique de la maladie de Pelizaeus-Merzbacher ne porte pas de mutation dans le gène PLP1. Ces patients sont reconnus comme ayant une maladie de type Pelizaeus-Merzbacher. Des mutations dans d’autres gènes (par exemple GJC2) ont été identifiées. Le terme Pelizaeus-Merzbacher-like disease (PMLD) est alors habituellement utilisé pour signifier la similarité de ces maladies, c’est-à-dire « like » en anglais.
Les mutations génétiques
Il existe différents types de mutations génétiques dont les principaux sont : 1) les duplications, 2) les mutations ponctuelles et 3) les mutations nulles.
- Dans le cas des duplications, un gène est présent en double exemplaire. Et en conséquence, la protéine issue du gène peut être produite en excès. Les duplications peuvent entrainer une augmentation de la fonction protéique, un « gain de fonction ».
- Dans les cas des mutations ponctuelles, il y a une erreur dans l’orthographe du gène. La protéine fabriquée peut alors être trop petite, mal fonctionner ou ne pas fonctionner du tout.
- mutations ponctuelles codantes : la mutation a une conséquence sur la composition de la protéine
- mutations ponctuelles non-sens: la mutation entraine la fabrication d’une protéine tronquée
- mutations ponctuelles non codantes : la mutation a une conséquence sur l’expression de la protéine, c’est-à-dire la quantité de protéine fabriquée
- Les mutations nulles ont pour effet d’empêcher toute production de la protéine du gène muté.
Les mutations génétiques
Les duplications : multiplication par deux
Depuis la découverte en 1989 révélant que les mutations du gène PLP1 entraînent la maladie de Pelizaeus-Merzbacher, il a été établi que la plupart des cas de Pelizaeus-Merzbacher sont dus à des duplications (ou plus rarement à des triplications ou même quintuplations) du gène PLP1 entier. En effet, les duplications sont retrouvées chez environ 50 à 75 % des familles touchées. Les duplications entraînent la forme classique de la maladie qui se manifeste tôt et présente souvent des symptômes sévères.
La taille et l’emplacement du fragment dupliqué sont variables d’une famille à une autre. Le gène PLP1 mesure environ 30 000 bases de long. Les plus petites duplications connues tournent autour de 100 000 bases d’ADN, alors que la plus importante mise en évidence à ce jour est de plus de 5 millions de bases. Le fragment d’ADN dupliqué peut donc être beaucoup plus grand que le seul gène PLP1. Il est envisagé que d’autres gènes soient également impliqués dans les différences neurologiques pouvant exister entre les familles, des gènes eux aussi dupliqués et situés avant ou après le gène PLP1 sur le chromosome X.
Il est actuellement estimé que la duplication a pour conséquence un surplus de fabrication de protéine. Les protéines accumulées sont toxiques pour les cellules appelées les oligodendrocytes qui fabriquent la myéline autour des axones des neurones.
Les mutations ponctuelles
Les mutations ponctuelles sont présentes chez 30 à 40% des patients atteints de la maladie de Pelizaeus-Merzbacher. Beaucoup de mutations ponctuelles de PLP1 ont été identifiées. La plupart de ces mutations ponctuelles sont uniques pour une famille. Et puisqu’elles sont uniques, il est difficile de prédire l’évolution de la maladie chez ces patients, particulièrement si aucun cas de la maladie n’avait été répertorié antérieurement dans la famille.
Les mutations non codantes
Récemment, on a trouvé des mutations non codantes dans une partie circonscrite du gène PLP1. Ces mutations entrainent une sous-expression relative de la protéine PLP1 par rapport à la protéine DM20 (une forme plus petite de la protéine PLP1). Elle est exprimée surtout dans le système nerveux périphérique et pendant certaines phases de myélinisation. L’IRM du cerveau est caractéristique. Elle montre une hypomyélinisation des structures normalement myélinisées assez tôt, d’où l’acronyme HEMS (Hypomyelination of Early Myelinating Structures).
Les mutations nulles
Pour finir, il existe des malades atteints de la maladie de Pelizaeus-Merzbacher pour lesquels le gène PLP1 est complètement absent ou bien avec une mutation située au début du gène et qui entraine une absence complète de production de la protéine. De façon surprenante, ces mutations, appelées mutations nulles, conduisent à un syndrome plus modéré que les duplications PLP1 ou que la majorité des mutations ponctuelles. Mais l’état des patients se détériore tout de même, et cette forme est moins bénigne qu’on ne le pensait initialement.
Conséquences des mutations
La sévérité d’une mutation dépend généralement de la façon dont la structure de la protéine est modifiée par cette mutation. Les mutations provoquant des modifications majeures dans la structure de PLP1 (ou un mauvais repliement de la protéine) entrainent la réponse pour protéine non repliée qui conduit à la mort des oligodendrocytes (voir encart).
La réponse aux protéines non repliées
Dans les cellules, les protéines sont fabriquées dans un compartiment appelé le réticulum endoplasmique. Quand la cellule en a besoin, comme c’est le cas lors des phases de croissance, la production de protéines s’intensifie.
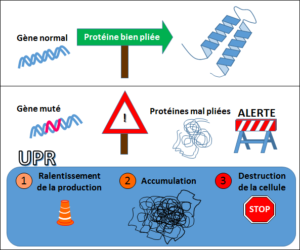
Quand la production est trop importante, la cellule commence à faire des erreurs, entrainant un stress du réticulum endoplasmique. Un signal d’alarme est alors tiré : une voie biochimique appelée la réponse aux protéines non repliées (la réponse UPR, pour Unfolded Protein Response en anglais), ralentit la chaine de fabrication des protéines pour permettre une amélioration de la qualité de la production. Cependant, quand le système est dépassé par la quantité de protéines mal formées, il peut conduire à la mort de la cellule pour éviter la survie d’une cellule inadaptée. C’est un système de protection naturel, un système qui tente dans un premier temps de réparer les dégâts, quitte à entrainer l’autodestruction de la cellule si la situation devient trop grave.
Quand un gène muté provoque un mauvais repliement de la protéine fabriquée, la cellule peut initier la réponse UPR conduisant à son autodestruction.
Cependant, quand le système est dépassé par la quantité de protéines mal formées, il peut conduire à la mort de la cellule pour éviter la survie d’une cellule inadaptée. C’est un système de protection naturel, un système qui tente dans un premier temps de réparer les dégâts, quitte à entrainer l’autodestruction de la cellule si la situation devient trop grave.
Quand un gène muté provoque un mauvais repliement de la protéine fabriquée, la cellule peut initier la réponse UPR conduisant à son autodestruction.
Les mutations ne modifiant que modérément la structure protéique, n’induisent pas autant de rétention de protéine dans la cellule et ne provoquent que peu ou pas de dégénérescence des oligodendrocytes. C’est le cas des mutations nulles, où la protéine PLP1 est totalement absente et non mal repliée. Il n’y a pas de toxicité liée au mauvais repli de la protéine.
Les symptômes de la maladie
La maladie de Pelizaeus-Merzbacher se caractérise par un nystagmus pendulaire, c’est-à-dire un mouvement oscillatoire involontaire des yeux, un tremblement de la tête et une hypotonie, mais aussi un retard de développement, une spasticité (contraction des muscles) et un déficit intellectuel variable. Le spectre clinique de la maladie est large, et on décrit 2 formes de la maladie suivant l’âge d’apparition et la gravité des symptômes : la forme classique de la maladie et la forme néonatale.
Les formes de la maladie
• PMD classique
La forme classique de la maladie de Pelizaeus-Merzbacher en est la forme la plus courante, et survient avant l’âge d’un an. Les premiers symptômes comprennent une faiblesse musculaire, des mouvements involontaires des yeux (nystagmus) et des retards de développement moteur au cours de la première année de vie. Ces retards du développement moteur et cognitif se manifestent à des degrés variables. Certains patients par exemple développent la possibilité de marcher de façon autonome, quand d’autres acquièrent le contrôle de la tête, mais sont dépendants d’un fauteuil roulant. De façon générale, le handicap moteur est plus sévère que le dysfonctionnement cognitif.
• PMD néonatale
La forme néonatale de la maladie en est la forme la plus grave : elle implique un retard de développement mental et physique et des symptômes neurologiques sévères. Les signes de la maladie peuvent être présents à la naissance ou apparaitre dans les premières semaines de vie. Ces enfants montrent un arrêt du développement à des étapes majeures telles que celle du contrôle de la tête et sont souvent alités pendant toute leur vie.
• Paraplégie spastique de type 2 (SPG2)
Ces patients représentent environ 20% des cas de la maladie.
Forme pure
La paraplégie spastique de type 2 est dite pure quand le seul phénotype présent est une paraplégie spastique, c’est-à-dire une paralysie plus ou moins complète des deux membres inférieurs associée à des spasmes, des convulsions, dus à une exagération du réflexe ostéo-tendineux [1]. Forme la moins sévère de la maladie de Pelizaeus-Merzbacher, les patients atteints de paraplégie spastique de type 2 pure ne présentent pas d’autres manifestations neurologiques.
[1] Exagération du réflexe ostéo-tendineux : résistance involontaire à un mouvement imposé, qui augmente avec la vitesse du mouvement.
Forme compliquée
Lorsque des caractéristiques neurologiques s’ajoutent à la paraplégie spastique, on parle de « paraplégie spastique de type 2 compliquée ». Ces caractéristiques neurologiques supplémentaires incluent un léger déficit intellectuel, une atrophie optique, un nystagmus et une ataxie apparaissant dans les premières années de vie. Les cas les plus modérés affichent la paraplégie spastique avec une déficience cognitive légère.
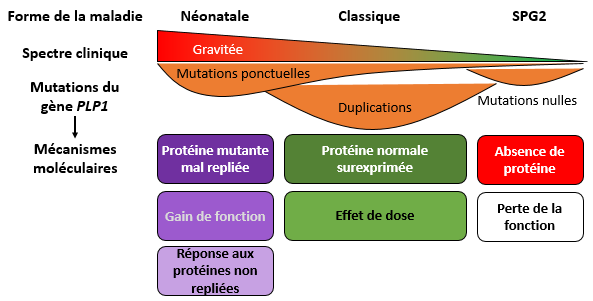
Les formes de la maladie de Pelizaeus-Merzbacher : conséquences des mutations génétiques
Néonatale : forme néonatale de la maladie de Pelizaeus-Merzbacher; Classique : forme classique de la maladie de Pelizaeus-Merzbacher; SPG2 : Paraplégie Spastique de type 2. PLP1 : protéine protéolipide 1. Gain de fonction : augmentation de l’activité normalement exercée par la protéine. Effet de dose : le niveau d’activité dépend de la dose de protéine formée. Perte de fonction : absence de l’activité normalement exercée par la protéine.
Adapté de Inoue Front Mol Biosci. 2017
La diversité des mutations responsables de la maladie de Pelizaeus-Merzbacher illustre l’équilibre délicat mis en œuvre par le programme génétique. Les protéines produites par chaque gène, comme la protéine PLP1, doivent être produites par chaque gène au bon endroit au bon moment, et en quantité ni trop grande, ni trop faible.
Le diagnostic de la maladie
La maladie de Pelizaeus-Merzbacher est évoquée devant le tableau clinique et les anomalies de la substance blanche à l’IRM. L’IRM montrera une hypomyélinisation complète (forme néonatale et quelques formes transitoires), partielle (pour la forme modérée) ou diffuse (maladie de Pelizaeus-Merzbacher, mutation non-sens de PLP1). L’étude des « potentiels évoqués auditifs du tronc cérébral[2] » peut être utile pour différencier la maladie de Pelizaeus-Merzbacher (absence d’ondes de II à V) de la maladie « PMD-like » (ondes II à V enregistrables). Un test génétique confirme le diagnostic.
[2] Le test des « potentiels évoqués auditifs du tronc cérébral » mesure la manière dont le cerveau traite les sons entendus.
Conseil génétique
Lorsqu’une mutation du gène PLP1 est identifiée dans une famille, il est possible d’examiner les membres de la famille pour la mutation et de fournir un diagnostic prénatal pour les parents ayant un risque de transmettre cette maladie. La maladie se transmet selon un mode récessif lié au chromosome X. Un garçon né d’une mère conductrice, a 50% de risque de présenter la mutation et de développer la maladie, tandis qu’une fille a 50% de risque d’être conductrice à son tour. Toutes les filles d’un homme affecté seront porteuses, mais aucun de ses fils ne sera affecté.
Le mécanisme simplifié conduisant à la maladie
Environ 75 % de la myéline est composée de graisses et de cholestérol, les 25 % restant étant des protéines. La protéine protéolipide 1 (PLP1), appelée aussi lipophiline, correspond à environ la moitié des protéines de la myéline et en est le constituant le plus abondant (mis à part les lipides). La protéine PLP1 est fabriquée à partir du gène PLP1, dans le réticulum endoplasmique des cellules, les oligodendrocytes, puis est incorporée dans la membrane de la cellule qui vient s’entourer autour des axones, les axes où circule l’information nerveuse. C’est ainsi qu’est formée la myéline.
Lorsqu’il y a une duplication du gène, la protéine est fabriquée en excès. La recherche sur les animaux a montré que l’excès de PLP1 s’accumule à l’intérieur de la cellule au lieu d’être orienté vers la membrane de la cellule pour y être incorporé dans la myéline. Les mutations ponctuelles ainsi que les autres petites mutations induisent généralement la substitution d’un des acides aminés par un autre, ou empêchent PLP1 d’être fabriquée sur toute sa longueur. Ceci a probablement comme conséquence une protéine ne pouvant pas se replier correctement ou une protéine ne pouvant plus interagir avec d’autres constituants de la myéline. Ces protéines mutantes sont toxiques pour les oligodendrocytes et les empêchent de produire une myéline normale.
PLP 1 représente normalement 50% du total des protéines présentes dans le système nerveux central, et seulement 1% dans les nerfs périphériques. Ceci explique pourquoi le système nerveux central est affecté par la maladie de Pelizaeus-Merzbacher, et non le système nerveux périphérique. Cependant, dans des cas exceptionnels (les mutations « nulles »), même la myéline des nerfs périphériques est affectée.
Comme pour les autres leucodystrophies hypomyélinisantes, la faible quantité de myéline fabriquée chez les patients atteints de la maladie de Pelizaeus-Merzbacher empêche l’acquisition des compétences plutôt qu’une perte de capacité.
Les traitements
La prise en charge de la maladie au quotidien
Actuellement, le traitement de la maladie de Pelizaeus-Merzbacher est un traitement symptomatique de soutien. Il peut inclure des médicaments contre la rigidité et la spasticité qui sont présentes chez la plupart des patients atteints après quelques années. En cas de convulsions ou d’épisodes apparentés à des convulsions, des antiépileptiques peuvent être nécessaires, bien qu’en général, l’épilepsie ne soit pas un événement fréquent.
La rééducation fonctionnelle est utile pour le maintien de la flexibilité des articulations, et maximise les capacités du patient. Les béquilles ou les déambulateurs peuvent aider à la marche. La chirurgie orthopédique peut également aider à réduire les contractures, les articulations bloquées par la spasticité ou une scoliose de la colonne vertébrale.
Si la parole ou la déglutition est altérée, un orthophoniste peut apporter des conseils importants. Quand la déglutition est sévèrement affectée, un tube d’alimentation, inséré directement dans l’estomac, peut aider à augmenter l’apport alimentaire. Des compléments de vitamine D et de calcium peuvent être utiles.
La prise en charge des patients atteints de la maladie de Pelizaeus-Merzbacher est multidisciplinaire et implique de nombreuses spécialités médicales. Le rôle des parents et des proches est essentiel.
Greffe de cellules souches neurales humaines
Un premier essai clinique a été mené pour évaluer la sécurité et l’efficacité de cellules souches neurales humaines dans le traitement de la maladie de Pelizaeus-Merzbacher. C’est un essai de thérapie cellulaire utilisant une banque de cellules souches. L’objectif de cet essai était d’injecter au patient malade, des cellules souches normales qui sauraient produire de la myéline, et d’en évaluer la sécurité.
Quatre jeunes enfants avec une forme précoce sévère de la maladie de Pelizaeus-Merzbacher ont reçu chacun 300 millions de cellules injectées dans chaque hémisphère cérébrale. Pour éviter un rejet immunitaire des cellules greffées, une immunosuppression a été administrée durant les 9 mois entourant la transplantation. Les patients ont été suivis pendant douze mois après la transplantation. Publiés fin 2012, les résultats indiquaient un bon profil d’innocuité, objectif de cette première étude. L’évaluation clinique avait néanmoins également révélé de faibles améliorations motrices et cognitives chez trois des quatre patients; le quatrième patient était resté cliniquement stable. De plus, les IRM suggéraient la fabrication minimale de myéline dans la région de la transplantation, persistant voire augmentant avec le temps. Ces quatre patients sont maintenant en phase de suivi à long terme.
Thérapie cellulaire utilisant les cellules du patient
Une nouvelle approche de thérapie cellulaire est aujourd’hui en développement. L’idée est de partir des cellules du patient lui-même, plutôt que des cellules étrangères d’un donneur. Cette approche fait appel à la technologie des cellules souches pluripotentes induites, iPSC (Voir ELA info 97).
A partir d’un prélèvement de peau ou de sang, les cellules du patient sont retransformées (induites) en cellules souches par stimulation de certains gènes. Elles sont ensuite corrigées en laboratoire pour ne plus porter la mutation responsable de la maladie de Pelizaeus-Merzbacher. Ces cellules ainsi corrigées pourraient ensuite être réinjectées chez le patient pour y fabriquer normalement de la myéline. Les premiers essais très encourageants ont été menés chez la souris et portent l’espoir de voir se mettre en place des essais cliniques et un traitement pour cette maladie orpheline.
ELA accompagne les cliniciens et les chercheurs travaillant sur la maladie de Pelizaeus-Merzbacher. Les recherches doivent continuer pour améliorer la compréhension de la pathogenèse de la maladie et le développement de traitements spécifiques et, idéalement, d’un remède. L’espoir se porte aujourd’hui vers la thérapie cellulaire et les cellules souches pluripotentes induites (iPSC) comme source de progéniteurs neuraux[3] dans le traitement de la maladie de Pelizaeus‐Merzbacher.
[3] Progéniteurs neuraux : cellules capables de se multiplier et de se transformer en tous types de cellules neurales, c’est à dire les neurones, les astrocytes ou encore les oligodendrocytes qui fabriquent la myéline.