PMD and other hypomyelinating leukodystrophies
Learn about Pelizaeus-Merzbacher disease (PMD)
Article reviewed by Prof. Nicole Wolf
Pelizaeus-Merzbacher disease (PMD) is a rare genetic disorder in the family of leukodystrophies. Its frequency is 1 per 100,000 births. It is one of the hypomyelinating leukodystrophies, white matter diseases characterised by a permanent deficit of myelin in the brain.
Pelizaeus-Merzbacher disease is named after two German doctors who described it in the early twentieth century. As early as 1885, one family had five boys with involuntary oscillatory eye movement, spasticity in their limbs, very limited head and body control and delayed cognitive development. Twenty-five years later, in 1910, re-examination of the family showed that 14 members of the family had the disease, including two daughters, and that all were descended from the same relative. It was also noted at that time that the disease was never passed from father to son, which has since been known to be a characteristic of genetic diseases in which the responsible gene is carried by the X chromosome.
Pelizaeus-Merzbacher disease presents in different forms depending on the age of onset of the first symptoms: a neonatal form, and a so-called “classic” form which occurs before the age of one. Two other less severe forms have been described: spastic paraplegia type 2 (which includes the recently described form, HEMS – Hypomyelination of Early Myelinating Structures) and the PLP1 null phenotype.
The gene whose mutation is responsible for Pelizaeus-Merzbacher disease is the PLP1 gene which is effectively located on the X sex chromosome (at Xq22.2). For this reason, men and women report the disease differently, and the disease typically affects boys or men. This gene codes for proteolipid protein 1 (PLP1): 188 disease-causing mutations have been described to date.
« PMD-like »
A small percentage of patients with the characteristic phenotype of Pelizaeus-Merzbacher disease do not carry a mutation in the PLP1 gene. These patients are recognised as having a Pelizaeus-Merzbacher-type disease. Mutations in other genes (e.g. GJC2) have been identified. The term Pelizaeus-Merzbacher-like disease (PMLD) is then usually used to signify the similarity of these diseases, hence the use of the word “like” in English.
Genetic mutations
There are different types of genetic mutations, the main ones being: 1) duplications, 2) point mutations and 3) null mutations.
- In the case of duplications, a gene is present in duplicate. And as a result, the protein from the gene can be produced in excess. Duplications can lead to an increase in protein function, a “gain of function”.
- In the case of point mutations, there is a spelling mistake in the gene. The protein being made may be too small, malfunctioning or not functioning at all.
- coding point mutations: the mutation has a consequence on the composition of the protein
- nonsensepoint mutations: the mutation results in the production of a truncated protein
- non-coding point mutations: the mutation has a consequence on the expression of the protein, i.e. the amount of protein produced
- Null mutations prevent any production of the mutated gene’s protein.
Genetic mutations
Duplications: doubling
Since the 1989 discovery that mutations in the PLP1 gene cause Pelizaeus-Merzbacher disease, it has been established that most cases of Pelizaeus-Merzbacher are due to duplications (or more rarely triplications or even quintuplications) of the entire PLP1 gene. Indeed, duplications are found in about 50-75% of affected families. Duplications result in the classic form of the disease which occurs early and often has severe symptoms.
The size and location of the duplicated fragment varies from one family to another. The PLP1 gene is about 30,000 bases long. The smallest known duplications are around 100,000 bases of DNA, while the largest identified to date is over 5 million bases. The duplicated DNA fragment may therefore be much larger than the PLP1gene alone. It is thought that other genes may also be involved in the neurological differences that may exist between families, genes that are also duplicated and located before or after the PLP1 gene on the X chromosome.
It is currently believed that duplication results in excess protein production. The accumulated proteins are toxic to the cells called oligodendrocytes that make myelin around the axons of neurons.
Point mutations
Point mutations are present in 30-40% of patients with Pelizaeus-Merzbacher disease. Many PLP1 point mutations have been identified. Most of these point mutations are unique to a family. And because they are unique, it is difficult to predict the course of the disease in these patients, especially if there has been no previous case of the disease in the family.
Non-coding mutations
Recently, non-coding mutations have been found in a limited part of the PLP1 gene. These mutations result in a relative under-expression of the PLP1 protein compared to the DM20 protein (a smaller form of the PLP1 protein). It is expressed mainly in the peripheral nervous system and during certain phases of myelination. The brain MRI is characteristic. It shows hypomyelination of normally myelinated structures at an early stage, hence the acronym HEMS (Hypomyelination of Early Myelinating Structures).
Null mutations
Finally, there are patients with Pelizaeus-Merzbacher disease in whom the PLP1 gene is completely absent or with a mutation at the beginning of the gene that results in a complete lack of protein production. Surprisingly, these mutations, called null mutations, lead to a milder syndrome than PLP1 duplications or the majority of point mutations. However, the patients’ condition deteriorates, and this form is less benign than initially thought.
Consequences of mutations
The severity of a mutation generally depends on how the structure of the protein is altered by the mutation. Mutations causing major changes in the structure of PLP1 (or misfolding of the protein) result in the unfolded protein response, which leads to oligodendrocyte death (see insert).
The response to unfolded proteins
In cells, proteins are made in a compartment called the endoplasmic reticulum. When the cell needs it, as it does during growth phases, protein production is intensified.
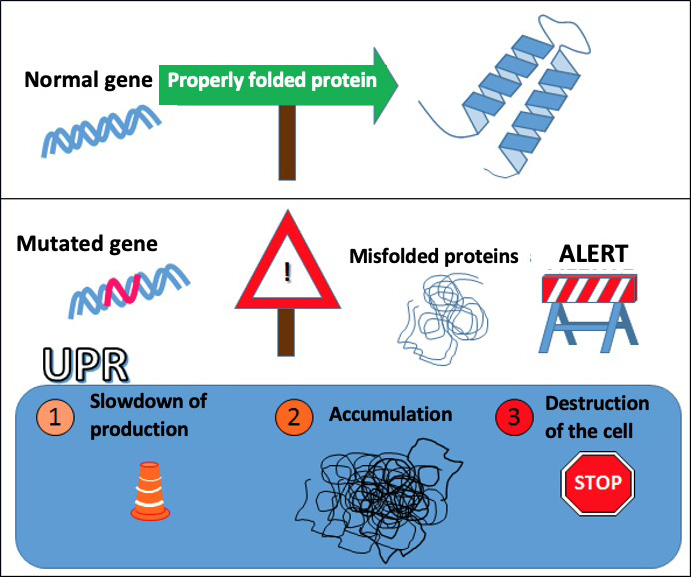
When the production is too high, the cell starts to make mistakes, causing stress to the endoplasmic reticulum. This raises a red flag: a biochemical pathway called the Unfolded Protein Response (UPR) slows down the protein manufacturing chain to allow for improved production quality.
However, when the system is overwhelmed by the amount of malformed proteins, it can lead to the death of the cell to prevent the survival of an unsuitable cell. It is a natural protection system, a system that first tries to repair the damage, even if it means the self-destruction of the cell if the situation becomes too serious.
When a mutated gene causes misfolding of the manufactured protein, the cell can initiate the UPR response leading to its self-destruction.
Mutations that only moderately alter the protein structure do not induce as much protein retention in the cell and cause little or no degeneration of oligodendrocytes. This is the case for null mutations, where the PLP1 protein is completely absent and not misfolded. There is no toxicity related to the misfolding of the protein.
Symptoms of the disease
Pelizaeus-Merzbacher disease is characterised by pendular nystagmus, i.e. an involuntary oscillatory movement of the eyes, head tremor and hypotonia, but also developmental delay, spasticity (muscle contraction) and a variable intellectual deficit. The clinical spectrum of the disease is wide, and 2 forms of the disease are described according to the age of onset and severity of symptoms: the classic form of the disease and the neonatal form.
Forms of the disease
• Classic PMD
The classic form of Pelizaeus-Merzbacher disease is the most common form and occurs before the age of one. Early symptoms include muscle weakness, involuntary eye movements (nystagmus) and delayed motor development in the first year of life. These motor and cognitive developmental delays occur to varying degrees. Some patients, for example, develop the ability to walk independently, while others gain head control but are dependent on a wheelchair. In general, motor disability is more severe than cognitive dysfunction.
• Neonatal PMD
The neonatal form of the disease is the most severe: it involves delayed mental and physical development and severe neurological symptoms. Signs of the disease may be present at birth or appear in the first few weeks of life. These children show developmental arrest at major milestones such as head control and are often bedridden throughout their lives.
• Spastic paraplegia type 2 (SPG2)
These patients represent about 20% of the cases of the disease.
Pure form
Spastic paraplegia type 2 is said to be pure when the only phenotype present is spastic paraplegia, i.e. more or less complete paralysis of both lower limbs associated with spasms, convulsions, due to an exaggeration of the tendon reflex[1]. The least severe form of Pelizaeus-Merzbacher disease, patients with pure spastic paraplegia type 2 have no other neurological manifestations.
[1] Exaggeration of the tendon reflex: involuntary resistance to an imposed movement, which increases with the speed of the movement.
Complicated form
When neurological features are added to the spastic paraplegia, it is called “complicated spastic paraplegia type 2”. These additional neurological features include mild intellectual deficit, optic atrophy, nystagmus and ataxia appearing in the early years of life. The most moderate cases display spastic paraplegia with mild cognitive impairment.
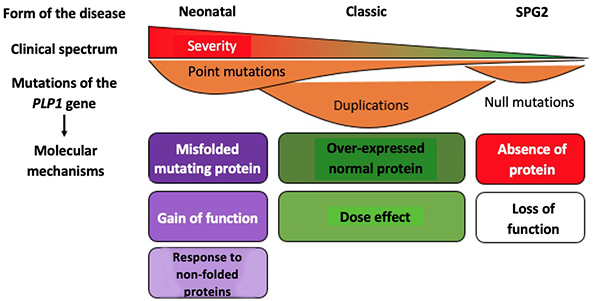
Forms of Pelizaeus-Merzbacher disease: consequences of genetic mutations
Neonatal: neonatal form of Pelizaeus-Merzbacher disease; Classic: classic form of Pelizaeus-Merzbacher disease; SPG2: Spastic paraplegia type 2. PLP1: proteolipid protein 1. Gain of function: increase in the activity normally performed by the protein. Dose effect: the level of activity depends on the dose of protein formed. Loss of function: absence of the activity normally performed by the protein.
Adapted from Inoue Front Mol Biosci. 2017
The diversity of mutations responsible for Pelizaeus-Merzbacher disease illustrates the delicate balance implemented by the genetic program. The proteins produced by each gene, such as PLP1, must be produced by each gene at the right place at the right time, and in quantities that are neither too large nor too small.
Diagnosis of the disease
Pelizaeus-Merzbacher disease is suggested by the clinical picture and white matter abnormalities on the MRI. The MRI will show complete hypomyelination (neonatal form and some transient forms), partial hypomyelination (for the moderate form) or diffuse hypomyelination (Pelizaeus-Merzbacher disease, nonsense mutation of PLP1). The study of “auditory brainstem responses”[1] can be useful to differentiate Pelizaeus-Merzbacher disease (absence of II-V waves) from “PMD-like” disease (recordable II-V waves). A genetic test confirms the diagnosis.
Genetic counselling
When a PLP1 gene mutation is identified in a family, it is possible to screen family members for the mutation and provide prenatal diagnosis for parents at risk of transmitting the disease. The disease is transmitted in an X-linked recessive mode. A boy born to a mother who is a carrier has a 50% chance of having the mutation and developing the disease, while a girl has a 50% chance of being a carrier in turn. All the daughters of an affected man will be carriers, but none of his sons will be affected.
The simplified mechanism leading to the disease
About 75% of myelin is composed of fats and cholesterol, and the remaining 25% is protein. Proteolipid protein 1 (PLP1), also known as lipophilin, makes up about half of the myelin proteins and is the most abundant component of myelin (apart from lipids). The PLP1 protein is made from the PLP1 gene in the endoplasmic reticulum of the cells, the oligodendrocytes, and is then incorporated into the cell membrane which surrounds the axons, the axons where nerve information travels. This is how myelin is formed.
When there is a duplication of the gene, the protein is made in excess. Animal research has shown that excess PLP1 accumulates inside the cell instead of being directed to the cell membrane for incorporation into myelin. Point mutations and other small mutations usually induce the substitution of one amino acid for another, or prevent PLP1 from being made along its entire length. This probably results in a protein that cannot fold properly or a protein that can no longer interact with other myelin components. These mutant proteins are toxic to oligodendrocytes and prevent them from producing normal myelin.
PLP 1 normally represents 50% of the total proteins present in the central nervous system, and only 1% in the peripheral nerves. This explains why the central nervous system is affected by Pelizaeus-Merzbacher disease, and not the peripheral nervous system. However, in exceptional cases (“null” mutations), even the myelin of peripheral nerves is affected.
As with other hypomyelinating leukodystrophies, the low amount of myelin produced in patients with Pelizaeus-Merzbacher disease prevents skill acquisition rather than loss of ability.
Treatments
Day-to-day management of the disease
Currently, treatment of Pelizaeus-Merzbacher disease is symptomatic and supportive. It may include medications for rigidity and spasticity, which are present in most patients after a few years. If seizures or seizure-like episodes occur, anti-epileptic drugs may be required, although epilepsy is not usually a frequent occurrence.
Rehabilitation is useful for maintaining joint flexibility and maximising the patient’s abilities. Crutches or walkers can help with walking. Orthopaedic surgery can also help reduce contractures, locked joints due to spasticity or scoliosis of the spine.
If speech or swallowing is impaired, a speech therapist can provide important advice. When swallowing is severely affected, a feeding tube, inserted directly into the stomach, can help increase food intake. Vitamin D and calcium supplements may be helpful.
The management of patients with Pelizaeus-Merzbacher disease is multidisciplinary and involves many medical specialties. The role of parents and relatives is essential.
Human neural stem cell transplantation
A first clinical trial was conducted to evaluate the safety and efficacy of human neural stem cells in the treatment of Pelizaeus-Merzbacher disease. This is a cell therapy trial using a stem cell bank. The objective of this trial was to inject the patient with normal stem cells that could produce myelin, and to evaluate their safety.
Four young children with a severe early form of Pelizaeus-Merzbacher disease each received 300 million cells injected into each cerebral hemisphere. To avoid immune rejection of the transplanted cells, immunosuppression was administered during the 9 months around the transplant. Patients were monitored for 12 months after transplantation. Published at the end of 2012, the results indicated a good safety profile, the objective of this first study. However, clinical evaluation also revealed small motor and cognitive improvements in three of the four patients; the fourth patient remained clinically stable. In addition, MRIs suggested minimal myelin production in the transplant area, persisting or even increasing over time. These four patients are now in long-term follow-up.
Cell therapy using the patient’s own cells
A new approach to cell therapy is now being developed. The idea is to use the patient’s own cells, rather than foreign cells from a donor. This approach uses induced pluripotent stem cell technology, iPSC (see ELA info 97).
From a skin or blood sample, the patient’s cells are transformed (induced) back into stem cells by stimulating certain genes. They are then corrected in the laboratory to no longer carry the mutation responsible for Pelizaeus-Merzbacher disease. These corrected cells could then be re-injected into the patient to produce normal myelin. The first very encouraging trials were conducted in mice and give hope that clinical trials and a treatment for this orphan disease will be set up.
ELA supports clinicians and researchers working on Pelizaeus-Merzbacher disease. Research must continue to improve the understanding of the pathogenesis of the disease, and the development of specific treatments and, ideally, a cure. Today, hope is focused on cell therapy and induced pluripotent stem cells (iPSCs) as a source of neural progenitors[3]in the treatment of Pelizaeus-Merzbacher disease.
[3] Neural progenitors: cells capable of multiplying and transforming into all types of neural cells, i.e. neurons, astrocytes or oligodendrocytes that produce myelin.