ALD/AMN
L’adrénoleucodystrophie
L’adrénoleucodystrophie liée au chromosome X (ALD) est une maladie génétique mondialement présente, rare et grave de la famille des leucodystrophies. Son incidence de 1 pour 17 000 naissances la place cependant comme la plus fréquente des leucodystrophies. C’est une maladie métabolique qui affecte non seulement le cerveau, la moelle épinière et les nerfs périphériques, mais aussi le cortex des glandes surrénales (et les testicules), une partie de son nom « adréno- » y fait référence.
L’adrénoleucodystrophie peut débuter dans l’enfance, l’adolescence ou l’âge adulte et se présenter sous de nombreuses formes cliniques de gravité variable :
• le plus souvent sous la forme d’une maladie lentement progressive de la moelle épinière et à un moindre degré des nerfs périphériques chez les hommes et les femmes : c’est l’adrénomyéloneuropathie (AMN) qui grève le pronostic fonctionnel moteur des membres inférieurs.
• plus rarement sous la forme d’une maladie rapidement évolutive du cerveau (ALD cérébrale) chez les garçons et les hommes adultes dont l’issue est le plus souvent fatale en l’absence de greffe de moelle osseuse.
1. La mutation génétique
Les chromosomes X et Y
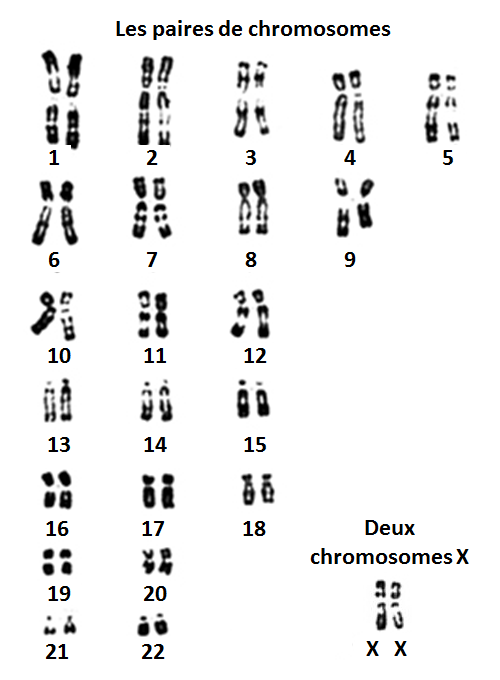 Les chromosomes sont des super-molécules présentes dans les noyaux des cellules. Ils contiennent l’ADN qui est le patrimoine génétique de tout individu.
Les chromosomes sont des super-molécules présentes dans les noyaux des cellules. Ils contiennent l’ADN qui est le patrimoine génétique de tout individu.
Chez l’homme, chaque cellule contient 46 chromosomes, regroupés en 23 paires. Une paire comprenant un exemplaire qui provient de la mère et un exemplaire qui provient du père. Une cellule dispose donc de deux copies de chaque gène, qui ne sont pas forcément identiques l’une à l’autre. Les chromosomes X et Y sont appelés chromosomes sexuels. Ils font partie de la même paire de chromosome, la 23ème paire. Leur nom vient du fait que les hommes et les femmes ne sont pas identiques vis-à-vis de cette 23ème paire. Les femmes ont 2 copies X et les hommes un X et un Y. Ainsi, une femme transmet toujours un X à ses enfants et un homme soit un X soit un Y, ce qui définira le sexe de l’enfant. Hommes et femmes ont donc tous le chromosome X en commun, mais les hommes n’en ont qu’une seule copie quand les femmes en ont deux. Les femmes n’ont pas de chromosome Y et n’ont donc aucune copie des gènes présents dans le chromosome Y à la différence des hommes.
e gène dont la mutation est responsable de l’ALD est le gène ABCD1 qui est situé sur le chromosome sexuel X (en Xq28). Les hommes, n’ayant qu’une copie du chromosome X, n’ont donc qu’une copie du gène ABCD1 et vont toujours subir les effets de la mutation si elle existe. Les femmes ont deux copies du chromosome X et donc deux copies du gène ABCD1 (l’une provenant de leur mère et l’autre copie provenant de leur père). Mais un seul des chromosomes X et donc un seul gène ABCD1 est fonctionnel dans les cellules des différents organes, et ce en proportion variable. Par exemple dans la moelle épinière, une femme conductrice hétérozygote peut avoir 80% des cellules qui expriment le gène ABCD1 normal ou à l’inverse 75% des cellules qui expriment le gène ABCD1 muté. Les femmes hétérozygotes ont toujours dans un organe donné au moins 10 à 20% des cellules qui expriment le gène ABCD1 normal. Ceci explique probablement pourquoi les femmes hétérozygotes ne développent jamais d’atteinte cérébrale et aussi pourquoi toutes ne développent pas des symptômes d’AMN à l’âge adulte.
![]()
Figure 1: la transmission génétique. Si la mère est conductrice d’une mutation sur le gène ABCD1 (partie gauche de la figure), pour chaque fille, il y a une chance sur deux qu’elle porte la mutation responsable de la maladie. Pour chaque fils, il a une chance sur deux qu’il porte la mutation et développe la maladie. Si le père est malade (partie droite), tous ses fils seront exempts de la maladie et toutes ses filles seront conductrices de la mutation responsable de la maladie.
2. Le mécanisme simplifié conduisant à la maladie
Le péroxysome
L’adrénoleucodystrophie est une maladie péroxysomale, c’est-à-dire qu’elle résulte du dysfonctionnement des peroxysomes qui sont des petites structures présentes dans toutes les cellules. Les péroxysomes sont chargés de dégrader les acides gras à très longues chaines (AGTLC). Le gène ABCD1 a été identifié en 1993 par les équipes du Pr Aubourg et du Pr Mandel. Il code pour la protéine ABCD1 (ou ALDP), un transporteur permettant l’entrée des acides gras à très longues chaines dans les péroxysomes où ils sont ensuite dégradés.
Dans l’adrénoleucodystrophie, la mutation du gène ABCD1 entraine un dysfonctionnement de ce transport et ne permet pas aux acides gras à très longue chaine d’entrer dans le péroxysome afin d’y être détruits par oxydation. C’est pourquoi ils s’accumulent dans toutes les cellules de l’organisme et dans le sang. On sait cependant aujourd’hui qu’une mutation du gène ABCD1 peut entraîner une anomalie de la fonction de certaines cellules sans que cela soit du à l’accumulation d’AGTLC.
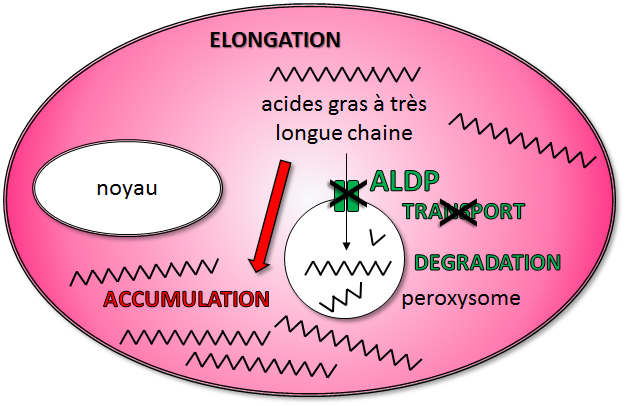
Figure 2: l’anomalie métabolique. Dans les cellules, les péroxysomes sont des compartiments dans lesquels les acides gras à très longues chaines (AGTLC) sont dégradés. La protéine ALDP, fabriquée à partir du gène ABCD1, permet de transporter les AGTLC dans les péroxysomes. Chez les patients atteints d’ALD, le gène ABCD1 est muté et la protéine ALDP qu’il code ne fonctionne pas : les AGTLC ne peuvent pas entrer dans les peroxysomes pour y être dégradés : ils s’accumulent dans les cellules complexés à différents autres lipides et protéines.
L’accumulation des acides gras à très longue chaine
Dans des conditions expérimentales en culture de cellules, les acides gras à très longue chaine semblent être toxiques pour les oligodendrocytes, les cellules qui fabriquent la myéline. Leur accumulation conduit à la mort cellulaire. Elle perturbe l’homéostasie du calcium et entraine le dysfonctionnement des mitochondries et la production d’espèces réactives de l’oxygène. Quand les oligodendrocytes meurent, la maintenance des gaines de myéline (qui sont en fait des prolongements des membranes des oligodendrocytes s’enroulant autour des axones) est altérée pouvant conduire à leur destruction, comme c’est le cas dans les formes cérébrales d’ALD. Dans l’AMN, l’accumulation d’AGTLC dans les oligodendrocytes de la moelle épinière ne conduit cependant pas à la mort des oligodendrocytes mais à un dysfonctionnement des axones sans démyélinisation significative. Lorsque les AGTLC s’accumulent dans la cellule, des acides gras à longue chaine plus courte (18 ; 20 ou 22 carbones) sont accessibles aux molécules chargées de les allonger, augmentant encore la quantité d’AGTLC. C’est un cercle vicieux.
La rupture de la barrière hémato-encéphalique
Récemment, il a été mis en évidence que la rupture de la barrière hémato-encéphalique, caractéristique de l’adrénoleucodystrophie visible par IRM, survient dans des régions du cerveau en avant de toute démyélinisation (Musolino et al., 2015). L’inflammation qui conduit à la dégradation de la myéline (par infiltration de leucocytes) semble être une conséquence directe de cette rupture de la barrière hémato-encéphalique, et non pas un phénomène directement lié à l’accumulation des AGTLC comme cela était envisagé précédemment. ABCD1 est très fortement exprimé dans les cellules endothéliales (et les astrocytes) de la barrière hémato-encéphalique. Cette équipe a montré que chez les patients ALD, et en absence d’ABCD1, des protéines, normalement situées au niveau des liaisons serrées et assurant l’étanchéité de la barrière hémato-encéphalique, ne s’y trouvaient pas correctement placées. Le passage est alors ouvert et les cellules et molécules de l’inflammation peuvent infiltrer le cerveau. L’absence de la protéine ABCD1 fonctionnelle pourrait être responsable de l’ouverture de la barrière hémato-encéphalique. Au niveau du cerveau, la rupture de la barrière hémato-encéphalique pourrait donc être en cause dans l’apparition de la forme cérébrale de l’adrénoleucodystrophie.
3. L’expression clinique
A la naissance, les garçons ou filles ne présentent pas de symptômes et l’expression clinique ultérieure est imprévisible. En revanche, l’ensemble des patients adultes hommes porteurs d’une mutation du gène ABCD1 finissent tous par développer des symptômes d’AMN avant l’âge de 55 ans, s’ils n’ont pas développé durant l’enfance une forme cérébrale. Comme indiqué précédemment, les filles ou femmes conductrices hétérozygotes ne développent jamais d’atteinte cérébrale. On sait cependant maintenant que 80% d’entre-elles finissent par développer des symptômes d’AMN lorsqu’elles atteignent l’âge de 60 ans.
Il n’existe pas de corrélation entre le génotype (le type de mutation du gène ABCD1) des patients et l’apparition des symptômes. En d’autres termes, au sein d’une même famille les garçons ou hommes adultes (frères, oncle-neveu par exemple) peuvent développer des manifestions différentes de la maladie. D’une famille à l’autre, même si la mutation du gène ABCD1 est identique dans les 2 familles, les patients atteints peuvent présenter des manifestations différentes de la maladie. Bien qu’ils ne soient pas encore précisément identifiés, des facteurs extérieurs semblent responsables du déclenchement du processus inflammatoire cérébral. Un traumatisme cérébral ou un accident vasculaire même mineur peuvent par exemple déclencher la démyélinisation cérébrale chez les patients ne présentant que des symptômes d’AMN ou même ne présentant encore aucun signe de la maladie. Des facteurs génétiques propres à chaque patient, et différents de la mutation du gène ABCD1, expliquent aussi une résistance à développer la forme la plus grave d’ALD (forme cérébrale démyélinisante) ou l’apparition des premiers signes d’AMN à un âge plus tardif ou sous une forme moins sévère.
La forme ALD cérébrale
Bien que tous les garçons atteints d’ALD naissent sans symptôme, la forme cérébrale de l’ALD se déclare le plus souvent entre 5 et 12 ans, jamais avant 4 ans. Des lésions de démyélinisation apparaissent d’abord sur les images d’IRM cérébrale et évoluent initialement lentement pendant plusieurs mois, voire années, sans le moindre symptôme identifiable. Et ce n’est que dans un deuxième temps qu’apparaissent simultanément des signes cliniques en rapport avec une progression importante et rapide des lésions de démyélinisation cérébrale. Sans greffe de moelle osseuse, et après une dégradation motrice et intellectuelle majeure, l’état des enfants peut se stabiliser pendant des années voire des décennies. Beaucoup cependant, surtout les plus jeunes, peuvent décéder de complications secondaires, respiratoires le plus souvent.
Les symptômes cliniques dépendent de la localisation des lésions de démyélinisation. D’une manière générale, la maladie évolue d’autant plus vite qu’elle débute tôt. Les symptômes initiaux les plus fréquemment observés sont : des difficultés scolaires, des troubles de l’attention, des difficultés pour se repérer dans l’espace, des difficultés du graphisme (dessin, écriture), des difficultés pour s’habiller (dyspraxie), des difficultés pour comprendre (ces garçons font souvent répéter les choses comme s’ils n’entendaient pas), des crises d’angoisses dues au fait qu’ils voient moins bien, comprennent moins bien et ne comprennent pas pourquoi.
Chez l’adulte, au moins 65 % des hommes entre 20 et 55 ans développent aussi une forme cérébrale visible à l’IRM cérébrale. Dans la moitié des cas, cette atteinte cérébrale se stabilise spontanément sans grande conséquence sur le plan moteur ou intellectuel et les patients peuvent mener une vie normale (sauf s’ils ont aussi des signes d’AMN, ce qui est presque toujours le cas). Dans l’autre moitié des cas, l’évolution des formes cérébrales de l’homme adulte est identique aux formes cérébrales du garçon, la période initiale d’aggravation lente des fonctions intellectuelles, visuelles et auditives étant cependant plus longue que chez les garçons. Tous les hommes adultes présentant une atteinte cérébrale démyélinisante présentent aussi des signes d’atteinte de la moelle épinière.
Certaines formes cérébrales n’évoluent pas immédiatement vers un stade inflammatoire (moins de 5 %). On parle alors de formes cérébrales chroniques dans lesquelles les lésions de démyélinisation évoluent très lentement. Ces patients peuvent être stables pendant plusieurs décennies. Ces patients ne développent pas de troubles visuels ou moteurs sauf lorsqu’une adrénomyéloneuropathie apparaît à l’âge adulte. Ils présentent cependant des déficits intellectuels importants qui compliquent leur adaptation à une vie scolaire ou professionnelle normale.
Les formes cérébrales ne sont pas décrites chez la femme.
L’adrénomyéloneuropathie
Les premiers symptômes de l’adrénomyéloneuropathie peuvent apparaitre chez l’homme entre 20 et 30 ans. Le plus souvent, ce sont des difficultés à courir, à marcher longtemps, en terrain accidenté. Le handicap moteur progresse lentement sur des décennies. L’adrénomyéloneuropathie est caractérisée par une paraparésie spastique (déficit moteur des membres inférieurs avec raideurs) associée à des troubles de l’équilibre (ataxie sensitive), des difficultés pour uriner. Une insuffisance surrénale, clinique ou simplement biologique, est très fréquente chez les hommes.
Les femmes conductrices de la mutation développent également les symptômes de l’adrénomyéloneuropathie. L’âge d’apparition est plus tardif, entre 40 et 50 ans. Les symptômes initiaux sont les mêmes que chez les hommes, mais les femmes sont plus souvent gênées par des troubles de l’équilibre que par une raideur des jambes, et ont plus souvent des douleurs neurogènes (à type de courant électrique, de broiement…) dans les jambes. Les difficultés urinaires sont aussi plus fréquentes et sévères et parfois associées à des difficultés de contrôle du sphincter anal.
Les symptômes de neuropathie périphérique sont rares, tant chez les hommes que chez les femmes hétérozygotes.
L’AMN évolue le plus souvent lentement sur des décennies avec des périodes de stabilisation apparente qui peuvent durer des années. Les membres supérieurs ne sont jamais atteints. Les fonctions intellectuelles ne sont pas touchées.
L’insuffisance surrénale (maladie d’Addison)
Environ 70 % des patients de sexe masculins atteints d’adrénoleucodystrophie finissent par développer à un moment de leur vie une insuffisance surrénale. Elle peut précéder les autres symptômes neurologiques de l’adrénoleucodystrophie (parfois pendant plusieurs dizaines d’années) ou bien survenir en même temps.
On peut la repérer à partir de l’âge de 3-4 ans par une pigmentation brune plus marquée au visage, au cou et au dos des mains, des cicatrices ou des plis de flexion des doigts, une fatigabilité accrue, des troubles digestifs, des nausées ou un manque d’appétit. L’insuffisance surrénale devrait être un signe évocateur d’une éventuelle adrénoleucodystrophie.
Chez les femmes conductrices ayant des symptômes d’adrénomyéloneuropathie, l’existence d’une insuffisance surrénale est très rare.
L’insuffisance testiculaire
Les hommes porteurs de la mutation présentent souvent des signes biologiques d’insuffisance testiculaire sans signes cliniques. Les hommes peuvent présenter des troubles de l’érection qui sont dus à l’atteinte de la moelle épinière. Très rarement, ils sont dus à l’insuffisance hormonale testiculaire qui entraîne une diminution de la testostérone et donc de la libido. Les femmes conductrices ne présentent aucune anomalie de la fonction ovarienne.
Les femmes conductrices :
Classiquement, l’adrénoleucodystrophie était considérée comme une maladie touchant uniquement les garçons et les hommes. Il est devenu clair depuis une quinzaine d’année qu’au moins 80% des femmes conductrices de la mutation responsable de la maladie finissent par développer des symptômes neurologiques d’AMN lorsqu’elles atteignent l’âge de 60 ans. Ces symptômes sont généralement moins sévères que chez les hommes adultes. Le pourcentage de femmes conductrices qui développent une forme de symptôme augmente graduellement avec l’âge, de 30% des femmes conductrices à l’âge de 20 ans, à plus de 80% des femmes après 60 ans.
4. Le diagnostic de la maladie
L’adrénoleucodystrophie est évoqué devant le tableau clinique neurologique ou endocrinien et les anomalies de la substance blanche à l’IRM cérébrale quand elles sont présentes. Elle est diagnostiquée par la mesure de la concentration des acides gras à très longue chaine dans le plasma. Trois valeurs sont mesurées : le taux d’acide gras saturés en 26 carbones, le rapport entre les taux d’acides gras saturés à 24 et 22 carbones, et le rapport entre les taux d’acides gras saturés à 26 et 22 carbones.
Ce diagnostic est totalement fiable chez les hommes. Chez les femmes hétérozygotes, le diagnostic est faussement négatif dans 20% des cas. Une analyse génétique (identification de la mutation du gène ABCD1) est indispensable pour confirmer ou infirmer le diagnostic.
Les changements biochimiques (augmentation de la concentration des acides gras à très longue chaine dans le plasma) surviennent avant les premiers symptômes, permettant le dépistage de l’adrénoleucodystrophie chez les garçons et filles nouveau-nés.
5. Le dépistage de la maladie dans chaque famille et le conseil génétique
Le dépistage dans chaque famille permet d’identifier des garçons encore asymptomatiques sur le plan neurologique et de proposer une greffe de moelle osseuse à un stade précoce de la maladie, s’ils développent une forme cérébrale. Pour cela, une IRM cérébrale doit être réalisée tous les 6 mois à partir de l’âge de 4 ans jusqu’à l’âge de 12 ans, puis une fois par an jusqu’à l’âge de 50 ans. Il permet aussi d’identifier des hommes adultes qui commenceraient à développer une forme cérébrale, ne présentant pas encore de symptômes évidents d’AMN, mais également d’identifier des patients de sexe masculin ALD, souffrant d’insuffisance surrénale qui, si non diagnostiquée et traitée à temps, peut s’avérer très grave et parfois mortelle.
Ce dépistage familial est aussi indispensable pour identifier toutes les femmes à risque d’être conductrices (hétérozygotes) de la mutation, et les informer du risque pour leurs futurs enfants et des modalités du diagnostic prénatal. Il l’est également pour les hommes ayant des filles en âge d’avoir des enfants, mais n’ayant pas encore développé la maladie (les premiers signes d’AMN peuvent parfois apparaître après l’âge de 45-50 ans chez les hommes).
Ce dépistage repose sur le dosage des AGTLC chez les hommes et la recherche de la mutation du gène ABCD1 chez les femmes.
6. Le dépistage néonatal
L’adrénoleucodystrophie peut aussi être dépistée dès 3 jours de vie chez tous les nouveau-nés à partir d’un prélèvement de sang déposé sur buvard (prélèvement de Guthrie). Des programmes de dépistage systématique de l’ALD sont mis en place aux Etats-Unis et bientôt aux Pays-Bas. Le dépistage néonatal de l’adrénoleucodystrophie, techniquement possible de nos jours, n’existe pas encore en France.
7. Les traitements
• Greffe allogénique de moelle osseuse
Une greffe allogénique de moelle osseuse peut être proposée pour le traitement des formes cérébrales précoces de la maladie. C’est le seul traitement qui permet, lorsqu’il est effectué au tout début de la maladie (en pratique quand les patients n’ont aucun symptôme), de stabiliser les lésions cérébrales de démyélinisation. Une greffe de moelle osseuse est inefficace, et peut même être néfaste, à un stade avancé ou tardif de la maladie. La greffe n’est possible que si un donneur compatible (moelle ou sang de cordon) est trouvé (voir le numéro précédent d’ELA infos). La greffe de moelle osseuse a la même efficacité chez les hommes adultes que chez les garçons. Chez les garçons, quand le donneur n’est pas un frère ou une sœur non atteint, le risque de mortalité dû aux complications de la greffe (rejet, réaction de greffon contre l’hôte) est de l’ordre de 12-13%. Pour les hommes, le risque de mortalité dû aux complications de la greffe est beaucoup plus élevé : de l’ordre de 35%, notamment à cause de complications bactériennes fatales que l’on n’observe plus chez l’enfant.
• Thérapie génique
La thérapie génique est un traitement encore en essai thérapeutique qui repose sur l’auto- greffe de cellules souches de moelle osseuse, prélevées chez les patients et réparées en laboratoire au moyen d’un vecteur viral de thérapie génique. Les premiers essais (4 patients dans un premier essai de phase I/II et 17 patients dans un deuxième essai de phase II/III) indiquent une très bonne efficacité de la thérapie génique, comparable à celle de la greffe de moelle osseuse, sans en avoir aucune des complications. Si les résultats de l’essai de phase I/II se confirment, la thérapie génique pourrait être relativement rapidement proposée à des garçons à un stade précoce de leur maladie cérébrale pour lesquels aucun donneur apparenté compatible (frère ou sœur non atteint) n’est disponible. La thérapie génique sera aussi proposée dans un second temps aux hommes adultes présentant une forme cérébrale évolutive potentiellement grave.
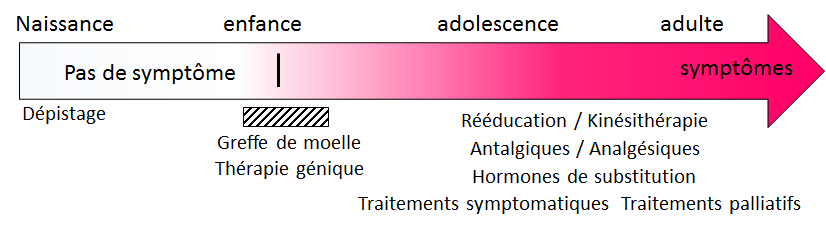
Figure 3: Evolution et traitements. À la naissance, les personnes atteintes d’ALD ne présentent pas de symptômes cliniques. Les acides gras à très longues chaines peuvent être dépistés à 3 jours de vie. Dès les tous premiers symptômes de la forme cérébrale de la maladie, une greffe de moelle osseuse peut être proposée. S’il n’y a pas de donneur compatible apparenté (frère ou sœur non atteints) disponible, une thérapie génique peut être envisagée. Une prise en charge symptomatique des patients et les traitements hormonaux de substitution sont possibles à tout moment. Une prise en charge psychologique et des traitements palliatifs peuvent être proposés.
• Traitement de l’adrénomyéloneuropathie
Le traitement de l’adrénomyéloneuropathie repose sur une prise en charge symptomatique des patients : rééducation motrice active et passive, traitement des troubles urinaires, de la spasticité, des douleurs des membres inférieurs, des troubles de l’érection. Le maintien d’une activité régulière bihebdomadaire de marche reste le « traitement » le plus efficace de l’AMN. A lui seul, il permet d’améliorer significativement le déficit moteur des membres inférieurs
• Traitement de l’insuffisance surrénale
Le traitement de l’insuffisance surrénale est possible via un traitement hormonal de substitution des corticostéroïdes surrénaliens (prescription d’hydrocortisone, voire de fludrocortisone). Ce traitement extrêmement simple, oral et quotidien, est indispensable et ne doit jamais être interrompu.
Huile de Lorenzo
Ce régime diététique repose sur une restriction alimentaire en acides gras à très longue chaîne et la prise d’huile enrichie en acide oléique et en acide érucique (« Huile de Lorenzo »). Il permet de normaliser en trois mois les taux plasmatiques d’acides gras à très longue chaîne, mais il n’a aucun effet dans les formes cérébrales de l’ALD et il n’y a pas de preuve scientifique d’un effet modificateur de l’huile de Lorenzo dans l’AMN ou même d’un véritable effet préventif. Enfin, il faut dire clairement que l’huile de Lorenzo est indiquée uniquement dans le traitement du défaut biochimique des ALD et ne joue aucun rôle dans d’autres pathologies neurologiques démyélinisantes ou progressives dans lesquelles les personnes peuvent métaboliser les acides gras à très longue chaîne.
• Traitement des formes cérébrales à un stade avancé
Des traitements palliatifs peuvent être proposés. Ils sont d’une importance cruciale pour améliorer la qualité de vie des enfants et des adultes atteints : lutte contre la douleur, la spasticité, traitement des complications orthopédiques, alimentation par sonde gastrique pour permettre un apport nutritionnel suffisant.
Malgré de nombreuses tentatives depuis plus de 30 ans, aucun traitement ne s’est avéré pouvoir stabiliser une forme cérébrale avancée d’ALD.
Diagnostic prénatal
Un diagnostic prénatal peut être proposé à toutes les femmes conductrices pour déterminer si l’enfant à naître est un garçon, et dans ce cas, s’il est porteur de la mutation du gène ABCD1 et donc à risque de développer durant l’enfance ou à l’âge adulte une des formes neurologiques de la maladie.
Ce dépistage nécessite un prérequis : que l’on ait bien démontré que la femme conductrice soit conductrice d’une mutation du gène ABCD1. Ce test nécessite simplement une prise de sang.
Les modalités du diagnostic prénatal sont les suivantes :
• dès le retard de règles, confirmation par un test de grossesse que la femme est bien enceinte et confirmation qu’elle est bien conductrice, faire très rapidement une échographie afin de dater exactement le début de la grossesse ;
• expliquer à la future maman, au couple, les risques d’avoir une fille conductrice, d’avoir un garçon atteint et, si c’est le cas, ce qu’on peut proposer aujourd’hui comme traitement ;
• vers 7-9 semaines de grossesse, on détermine par une simple prise de sang si la future maman attend un garçon ou pas. Le test repose sur l’identification de séquences du chromosome Y dans le sang maternel (il y a toujours des cellules du fœtus qui passent dans le sang des femmes enceintes). Ce test est totalement fiable ;
• entre 11 et 13 semaines de grossesse, on fait une ponction de trophoblastes. Cela revient à prendre un tout petit bout du placenta qui a la même origine génétique que le fœtus. Cette ponction comporte un risque minime d’avortement spontané ;
• on fait ensuite la recherche de la mutation du gène ABCD1 sur ce prélèvement de trophoblaste. Le résultat est habituellement donné 8-10 jours après ;
• beaucoup plus rarement, en pratique quand le diagnostic de la grossesse a été fait tard ne permettant plus de proposer à temps une ponction de trophoblaste, on fait entre 15 et 18 semaines de grossesse une ponction amniotique. Celle-ci permet de recueillir des cellules du fœtus sur lesquelles on fait : a) un caryotype (examen des chromosomes) afin de déterminer ci c’est un garçon ; b) et si c’est le cas, une recherche de la mutation du gène ABCD1.
Si le garçon à naître est porteur de la mutation du gène ABCD1, on peut proposer à la femme, au couple, une interruption volontaire de grossesse.
Le diagnostic préimplantatoire (implantation d’un œuf obtenu par fécondation in vitro après avoir vérifier que cet œuf composé de quelques cellules ne présente pas une mutation du gène ABCD1) est en pratique aujourd’hui réservé aux femmes qui ont déjà fait au moins une, voire deux interruptions volontaires de grossesse suite à un diagnostic prénatal. La raison en est simple : il existe très peu de centres compétents pour faire ce diagnostic en France, cette procédure compliquée n’est pas sans risque et l’ALD n’est qu’une maladie rare parmi beaucoup d’autres. Il y a beaucoup d’autres demandes d’autres couples concernés par d’autres maladies rares.
• Prise en charge psychologique
Une prise en charge psychologique doit accompagner non seulement les patients, mais aussi les frères et sœurs des patients, leurs parents, le conjoint et bien souvent plusieurs membres de la même famille.
• Prise en charge de la maladie au quotidien
Un soutient de kinésithérapie ainsi que la gestion des complications urologiques peuvent être proposés pour la gestion des troubles liés à l’adrénomyéloneuropathie, pour permettre le maintien d’une qualité de vie personnelle et professionnelle.
Conclusions
Plusieurs zones d’ombre existent encore pour l’ALD tant du point de vue de la compréhension de certains mécanismes (pourquoi les 2/3 des patients de sexe masculin développent une atteinte cérébrale soit dans l’enfance soit à l’âge adulte et les autres non…) que du traitement des formes cérébrales avancées.
Mieux connaitre la maladie a permis de développer un test qui pourrait identifier tous les garçons nouveau-nés, permettant de les suivre très tôt et de leur offrir un traitement dont on sait aujourd’hui qu’il n’est efficace que précocement réalisé. Ce dépistage néonatal non effectué en France sera un nouveau combat pour ELA. Il permettrait d’éradiquer les formes cérébrales avancées pour lesquelles on ne peut toujours rien faire.