Aicardi-Goutières syndrome
Aicardi-Goutières syndrome
In 1984, Jean Aicardi and Françoise Goutières, two French pediatric neurologists, described a genetic brain disease beginning in childhood that mimics the characteristics of viral infections that children suffer from in the womb.
Clinical indicators of this disease, now known as Aicardi-Goutières syndrome (AGS) include:
- Calcium build-up (calcification) in the brain, best observed by CT scan
- Changes in the white matter of the brain and spinal cord, best seen by MRI
- High levels of white blood cells, alpha interferon, and pterins (proteins produced by the body to fight a viral infection) in the cerebrospinal fluid (which can be tested by lumbar puncture)
- Distinctive frostbite-like lesions on the hands and feet, usually more severe in the cold
Genetic
Aicardi-Goutières syndrome is usually an inherited genetic disorder with autosomal recessive inheritance. This means that for a couple with an affected child, there is a 1 in 4 risk of having a sick child with each pregnancy. Three cases were identified with transmission of the “new dominant” type. In these rare cases, the risk of recurrence is very low.
From:
Dr, Adeline Vanderver
Program Director of the Leucodystrophy Center of Excellence
Children’s Hospital of Philadelphia
Philadelphia, PA 19104 – USA
March 2018
Anticipatory Guidance in AGS
Aicardi Goutieres Syndrome (AGS) is a genetically heterogeneous disorder that can be caused by mutations in a series of genes with a final endpoint of upregulation of the IFN pathway and the innate immune system. In some genotypes (TREX1, RNASEH2A,B,C and SAMHD1) it is assumed that accumulation of endogenous retroelements through deficiency of AGS proteins triggers the endogenous RNA or DNA sensing mechanisms and downstream IFN activation. In other genotypes (IFIH1 and ADAR1) it is thought that direct IFN activation occurs.
There is no current cure for AGS, though clinical trials are anticipated.
However, much can be done for symptomatic management of AGS at this time. In addition to the above patient specific recommendations, careful attention should be paid to the following medical complications:
Encephalopathy/irritability and sleep: Many children with AGS suffer from significant irritability. In some cases, medications to address this can be helpful, providing the opportunity for sleep in particular. Medications that have been previously tried in our patient population with some success, in particular at bedtime include: melatonin, clonazepam and guanfacine. These should be carefully titrated to minimize sedation and daytime sleepiness. In addition, irritability may wane over time and when no longer needed, these medications should be weaned to avoid the complications of polypharmacy. Patients with AGS may develop sleep apnea (obstructive and or central) and this complication should be sought on medical history.
Recurrent aseptic febrile illnesses: patients with AGS may have recurrent events of fever, sometimes associated with increased irritability, rash and even on occasion documented elevated cerebrospinal fluid white blood cells. In most cases these are not associated with a provoking viral or bacterial illness though care should be taken to exclude this.
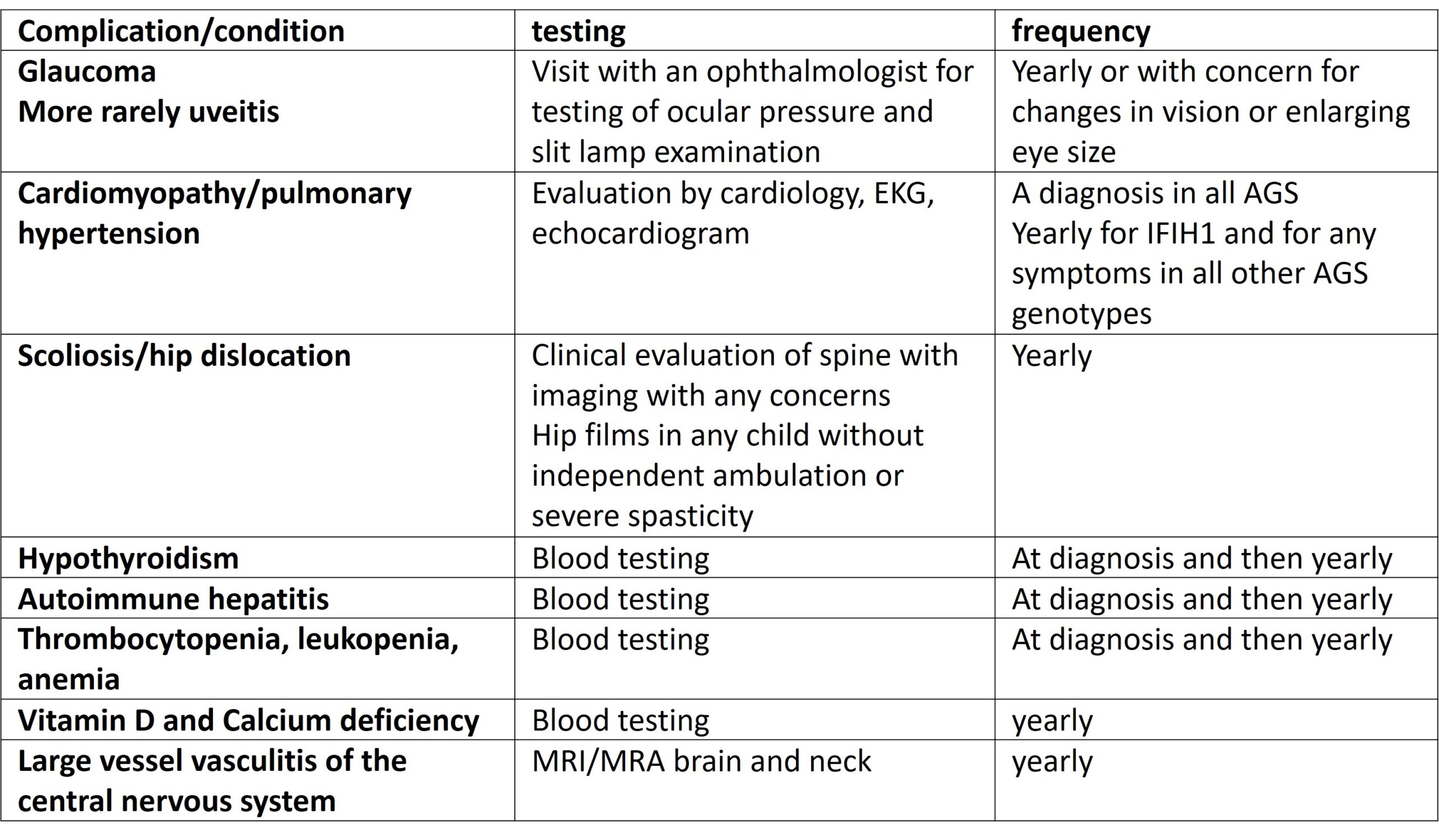
Eyes: Glaucoma is a known complication of AGS and should be sought at yearly ophthalmologic evaluations. Decreased blinking as a result of encephalopathy should be looked for a treated with artificial tears to prevent corneal injury. Vision loss related to brain injury should be considered (cortical visual blindness) and managed in a rehabilitation environment.
Dental: dental root resorption and gingival inflammation has been seen in AGS, particularly in IFIH1 associated AGS; regular dental care is recommended every 3 months
Swallowing: Patients with AGS will often have impaired swallowing. Careful monitoring for aspiration risk and management of hypersalivation should be considered.
Respiratory: children with AGS may have respiratory complications related to their underlying brain injury. A yearly influenza vaccination should be considered in all patients with AGS. Patients should be followed by a pulmonologist on an at least yearly basis.
Cardiac: occasional patients will have enlarged hearts for reasons not fully understood, but thought to relate to immune injury of the heart muscle. Other patients may have other kinds of inflammatory heart and valve changes. This potentially serious complication should be followed with a cardiologist on a yearly basis. Additionally, some individuals, in particular those with IFIH1 variants may be susceptible to pulmonary hypertension, and this complication should be sought on yearly echocardiograms and electrocardiograms.
Nutrition: Alternate nutrition via G tube is often necessary in patients with AGS. No particular dietary needs exist, although in rare case, inflammatory bowel disease can complicate meeting nutritional needs.
Gastrointestinal disease is common in AGS and can include reflux, which is often diagnosed in infancy and often initially blamed for the extreme irritability seen in AGS patients. Additionally, patients frequently suffer from constipation and an initial over the counter approach is the use polyethylene glycol (Miralax) which can be titrated to produce a soft stool daily. More rarely individuals with AGS can have inflammatory bowel disease and this should be considered in patients with bloody stools.
Many individuals with AGS may have moderately elevated AST, ALT or GGT. In many cases these are stable and without any clinical manifestations. Some severely affected patients will have a disorder that resembles autoimmune hepatitis. Patients with AGS should be followed by a gastroenterologist on a yearly basis, and consideration given to laboratory testing of liver related parameters.
Endocrine: patients with AGS may develop thyroid disease and yearly thyroid testing is recommended. More rarely, individuals may develop diabetes and diabetes insipidus and symptoms such as increased urination and recurrent dehydration should be evaluated for these disorders.
Hematologic: some patient with AGS will have low platelets and anemia, typically as newborns. This may be associated with hepatosplenomegaly. This complication usually does not persist. However, in some cases anemia, thrombocytopenia or leukopenia can develop later, and if any suggestive symptoms arise, a complete blood count is indicated.
Orthopedic: children affected by AGS can occasionally develop autoimmune joint disease and contractures. These conditions should be managed in consultation with a rheumatologist. More typically joint damage occurs as a late complication of spasticity and dystonia. Yearly hip films are recommended to assess for hip subluxation/dislocation. Scoliosis should be assessed as suggested by clinical evaluation. Repair of hip dislocation and scoliosis should be considered in the context of the overall health of the child but should take into consideration that children with AGS are likely to live for many years. Decreased mobility and multiple medications may also result in osteopenia and at least yearly vit D and Calcium measurements with appropriate replacement should be considered.
Skin: skin manifestation can be a painful aspect of AGS. Chilblains should be prevented where possible by minimizing exposure to cold weather and pressure. There are limited effective treatment options for the skin complications of AGS. On occasion, more severe and extensive skin manifestations can be seen, such as paniculitis, or extensive rashes. These maybe very uncomfortable and should be managed with a dermatologist or rheumatologist.
Spasticity/Dystonia: patients with AGS can have complex motor impairments including spasticity and dystonia and may benefit from a medical approach to these symptoms to improve comfort and functional status or positioning. Medications used safely and effectively in AGS include baclofen and trihexiphenidyl . Some parents have reported decline in function after use of botulinum toxin and other children have tolerated this very well. If botox is not used, this does not preclude the use of phenol to the larger muscles of the lower extremities to improve comfort and function.
Seizures: seizures are typically easily controlled and fairly uncommon in AGS. Patients with AGS have many non seizure abnormal movements related to basal ganglia injury and if possible, events should be characterized with EEG monitoring prior to treatment with anticonvulsants.
Autonomic dysfunction: over time, many patients with AGS will develop autonomic dysfunction. This may include episodes of sweating, fast heart rates and heavy breathing and temperature changes. This may also affect coloration of the extremities (alternating red and purplish discoloration) or changes in temperature of the hands and feet. Some medications can be used to limit these events if they are disruptive to the individual’s quality of life.
Developmental delay: patients with AGS will often have severe developmental delay. Nevertheless, care should be taken not to underestimate the abilities of a child with AGS and rehabilitative measures such as augmentative communication evaluations and regular evaluations in a multidisciplinary rehabilitation team should be considered.
Neurologic: in addition to the above, individuals with SAMHD1 associated AGS are at risk for Moya Moya disease, and large vessel vasculitis. Yearly MRI of the brain with MRA is recommended in this subgroup of individuals.