LTBL
Le syndrome de leucoencéphalopathie-anomalies du thalamus et du tronc cérébral-hyperlactatémie (ou LTBL)
Le syndrome de leucoencéphalopathie-anomalies du thalamus et du tronc cérébral-hyperlactatémie (ou LTBL) est une maladie génétique extrêmement rare de la famille des leucodystrophies. Sa prévalence est de moins d’une personne affectée sur 1 000 000. Elle fait partie des leucodystrophies hypomyélinisantes, ces pathologies de la substance blanche caractérisées par un déficit permanent de myéline au niveau du cerveau.
Le syndrome étant si rare, la maladie est mal connue et très peu de d’informations sont disponibles.
Le syndrome LTBL se manifeste dès la petite enfance. Il se caractérise par des anomalies dans certaines régions du cerveau, y compris dans le thalamus et le tronc cérébral (partie du cerveau qui se connecte à la moelle épinière), et par un niveau élevé d’une substance appelée lactate dans le cerveau et dans tout l’organisme. Le syndrome entraine généralement des problèmes moteurs et de contrôle de la fonction musculaire.
Le gène dont la mutation est responsable du syndrome LTBL est le gène EARS2 qui est situé sur le chromosome 16 (en 16p12.2). Ce gène code pour une protéine, la glutamyl-ARNt synthétase impliquée dans la fabrication des protéines dans les mitochondries.
Fabriquer des protéines : une industrie !
Dans les cellules, toute une industrie est responsable de la fabrication des protéines. Ces molécules, diverses et nombreuses dans la cellule, assurent une multitude de rôles. Les protéines sont toutes fabriquées selon le même principe, dans les mêmes usines, mais en suivant les instructions spécifiques à chacune qui sont contenues dans le génome sous la forme de séquence d’ADN.
Dans le noyau des cellules se cache le génome, véritable bibliothèque archivant les recettes de fabrication de toutes les protéines. A partir de ces instructions, un exemplaire de la recette sera recopié sous forme d’ARN, pour être transmis à l’unité chargée de la production. Des unités de production fabriquent alors les protéines en assemblant les blocs de construction en suivant les instructions recopiées dans l’ARN.
USINE DE PRODUCTION
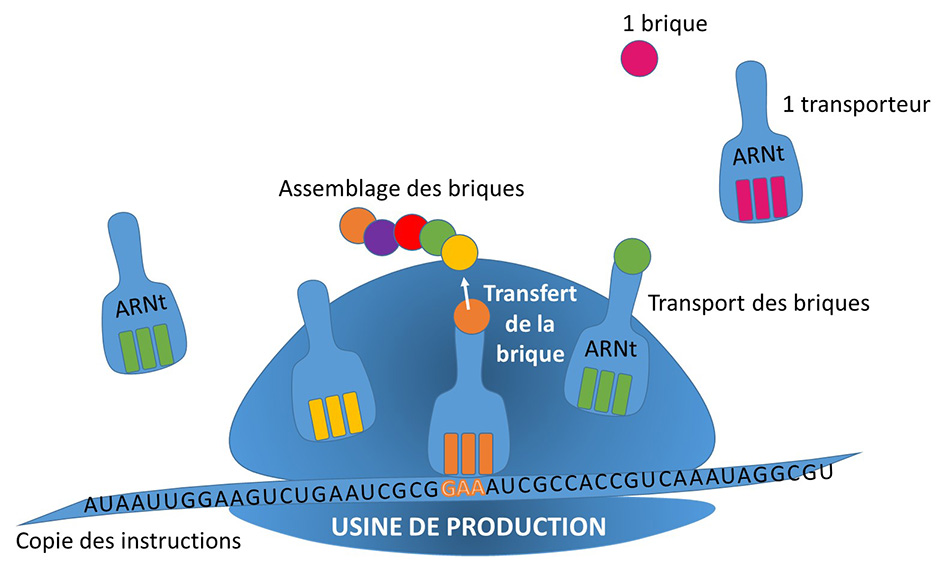
L’ARNt tient son petit nom de sa fonction de transfert des blocs (les acides aminés) nécessaires à la construction des protéines. C’est ce transporteur qui apporte le matériel dans la zone de production, puis permet son rattachement aux briques déjà assemblées.
L’ARNt synthétase est chargée de fixer la bonne brique sur l’ARNt. D’elle dépend la préparation de cet ouvrier spécialisé et donc toute la chaine de production en général.
Les symptômes de la maladie
C’est en 2012 qu’une équipe de chercheurs a pu mettre son nom sur le gène qui est muté dans le syndrome de LTBL. Pour cela les chercheurs ont analysés, par séquençage d’exome de nouvelle génération, l’ADN de 12 patients présentant les mêmes caractéristiques. La maladie est tellement rare que pour réaliser ce travail, les petits patients venaient de tous les coins du monde : un italien, deux belges, deux anglais, un américain, un israélien, un suisse, un portugais, un allemand, et deux brésiliens.
Les formes de la maladie
Les patients atteints du syndrome de LTBL présentent tous une apparition des symptômes à un stade infantile et une progression rapide de la maladie avec des anomalies sévères visibles en imagerie par résonance magnétique et une augmentation du taux de lactate. La maladie se présente sous deux formes : une forme légère et une forme sévère.
Dans sa forme légère, à partir de 6 mois d’âge, la maladie entraine une perte de capacités mentales et de mouvement (régression psychomotrice). Une raideur musculaire (spasticité) et l’extrême irritabilité sont courantes, et certaines personnes développent des convulsions. Les patients se rétablissent ensuite partiellement. Les étapes de développement peuvent être franchies avec du retard, mais les enfants sont capables d’acquérir de nouvelles capacités dans les années qui suivent. L’imagerie par résonance magnétique présente des améliorations frappantes et les taux de lactate baissent.
Pour les patients présentant la forme sévère de la maladie, les symptômes commencent peu après la naissance. Ces nourrissons ont généralement un développement retardé des capacités mentales et de mouvement (retard psychomoteur), un tonus musculaire diminué (hypotonie), une tension musculaire involontaire (dystonie), une spasticité musculaire et des convulsions. Certains ont des niveaux extrêmement élevés de lactate (acidose lactique), ce qui peut causer de graves problèmes respiratoires et cardiaques. Une insuffisance hépatique survient chez certains nourrissons gravement atteints. On note par la suite une stagnation clinique, une atrophie cérébrale visible à l’IRM et une augmentation persistante du taux de lactate.
Le diagnostic de la maladie
Le diagnostic peut aujourd’hui être évoqué par le tableau clinique du patient, et l’image caractéristique par IRM. Une élévation de lactate est également mesurable et indicative. Pour confirmer le diagnostic, le séquençage du gène EARS2 montrera une mutation sur chacune des deux copies du gène (maladie autosomale récessive : les parents portent chacun une copie du gène muté, mais ils ne montrent généralement pas les signes et les symptômes de la maladie).
Le mécanisme simplifié conduisant à la maladie
La mitochondrie est une structure à part dans la cellule, chargée d’assurer la respiration de la cellule, et fabriquant ainsi de l’énergie pour la cellule. Pour assurer ses activités propres, la mitochondrie utilise son propre système de fabrication des protéines, sa propre usine. Mais le principe reste le même (voir schéma : Fabriquer des protéines : une industrie !). L’ARNt synthétase issue du gène EARS2, permet la fixation du glutamate (la brique) sur son ARN de transfert qui travaille dans les mitochondries.
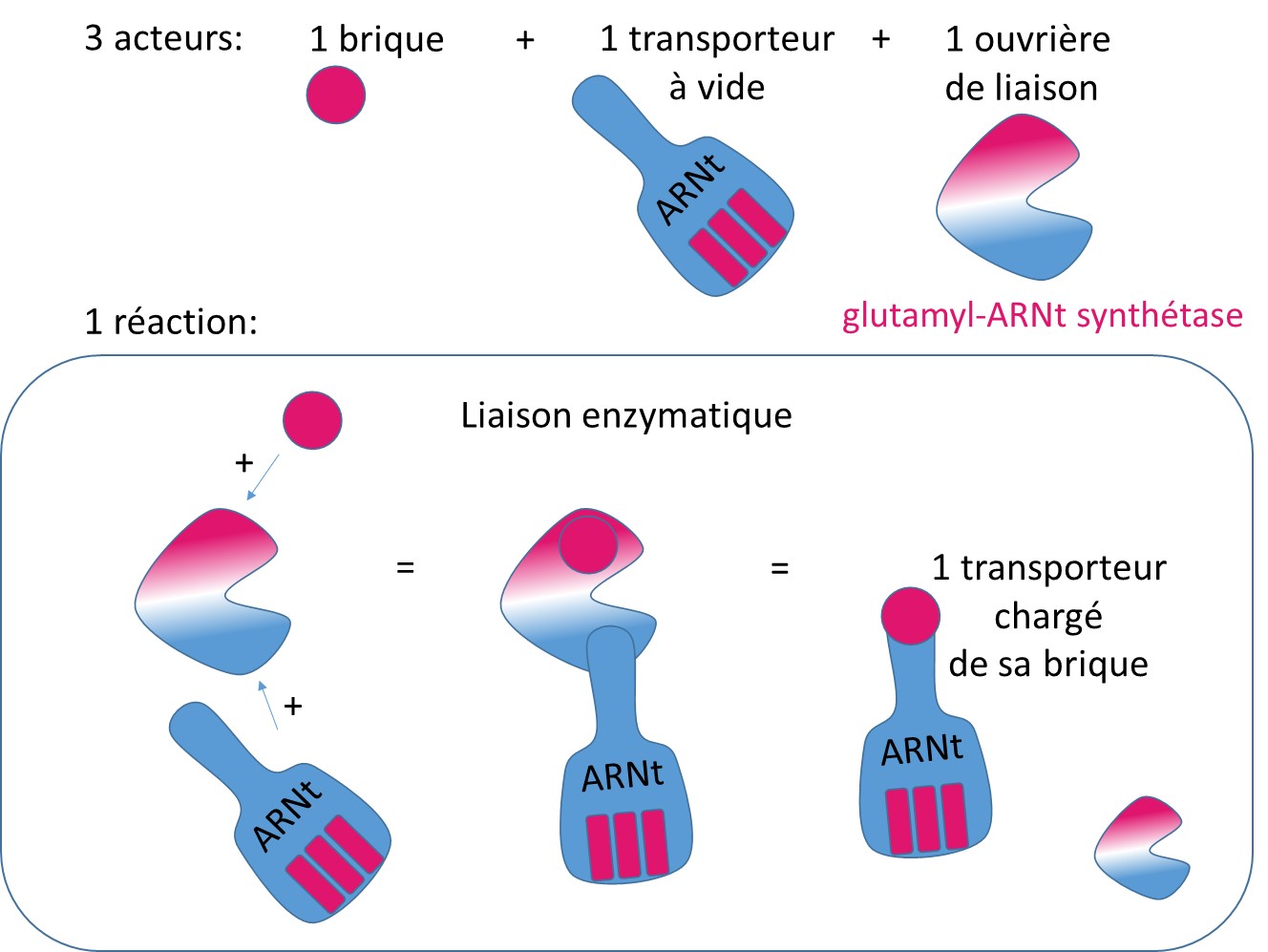
La liaison enzymatique des acides aminés sur les ARNt : la glutamyl-ARNt synthétase, issue du gène EARS2, est l’enzyme (l’ouvrière) nécessaire à l’assemblage de l’acide aminé glutamate (la brique) sur l’ARNt (le transporteur).
Dans le syndrome LTBL, les chercheurs ont pu identifier le gène qui est muté, et estiment que la quantité de glutamyl-ARNt synthétase est réduite chez les patients. La quantité réduite d’enzyme empêche probablement l’assemblage normal de nouvelles protéines dans les mitochondries. Il est supposé que cela perturbe la production d’énergie par la mitochondrie. Cependant, on ne sait pas exactement aujourd’hui comment les mutations du gène EARS2 conduisent aux caractéristiques cliniques du syndrome LTBL.
Les traitements
La prise en charge de la maladie au quotidien
Le traitement est symptomatique et se conduit idéalement dans un cadre multidisciplinaire par des spécialistes expérimentés dans le soin des personnes atteintes de leucodystrophies. Des médicaments sont proposés pour gérer le tonus musculaire. La kinésithérapie intensive peut être utilisée pour améliorer la mobilité et la fonction. Le traitement de l’ataxie, des crises épileptiques et des problèmes cognitifs suit les standards habituels, selon les besoins de l’individu.
Une surveillance doit être maintenue pour évaluer la croissance et l’état nutritionnel de l’enfant. Des examens physiques et / ou radiographiques en série des hanches et de la colonne vertébrale peuvent permettre de surveiller les complications orthopédiques. L’historique est faite des signes et des symptômes de crises.
Quand une maladie est si rare, comment espérer s’y retrouver et mettre un nom sur ce qui touche votre enfant. Madame Karine Garest, mère de Marion, nous raconte comment elle a pu enfin le faire en écoutant la description de la maladie faite par le Pr. van der Knaap lors d’un colloque familles / chercheur ELA.
« En 2012 lors du colloque organisé par ELA je me suis rendue à l’atelier sur les leucodystrophies indéterminées, le Pr. van der Knaap présentait une nouvelle forme de leucodystrophie, la LTBL. Lors de la description elle faisait part d’une attaque de lactate au niveau du cerveau entre 0 et 2 ans qui détruisait plus ou moins la myéline, et qu’on retrouvait un taux de lactate important dans le sang. Je me suis alors souvenu d’une prise de sang que Marion avait faite aux alentours de ses 9 mois et où on avait un taux de lactate élevé. Ceci a commencé à me renvoyer à notre histoire. Ensuite elle a fait part de l’évolution des enfants en disant qu’après cette attaque il n’y avait plus de régression et qu’au contraire les enfants progressaient et c’était le cas de Marion puisqu’elle a tenu assise à 2 ans, a marché à 5 ans… donc j’ai reconnu Marion dans ce descriptif et nous avons pu échanger à la fin de l’atelier et échanger par mail par la suite jusqu’au dépistage sanguin qui a confirmé que Marion souffrait de cette forme de leucodystrophie…
Pour l’instant elle est la seule diagnostiquée en France, ce qui, comme nous a dit le Pr. Wolf au colloque, n’est pas possible, il y a certainement d’autres personnes atteintes de cette forme de leucodystrophie, alors j’espère que mon témoignage permettra à d’autres familles de mettre un nom sur la maladie de leur enfant car si je n’avais pas été au colloque cette année-là Marion ferait toujours partie des leucodystrophies indéterminées… »