Les leucodystrophies
Les leucodystrophies : des maladies qui touchent particulièrement les enfants
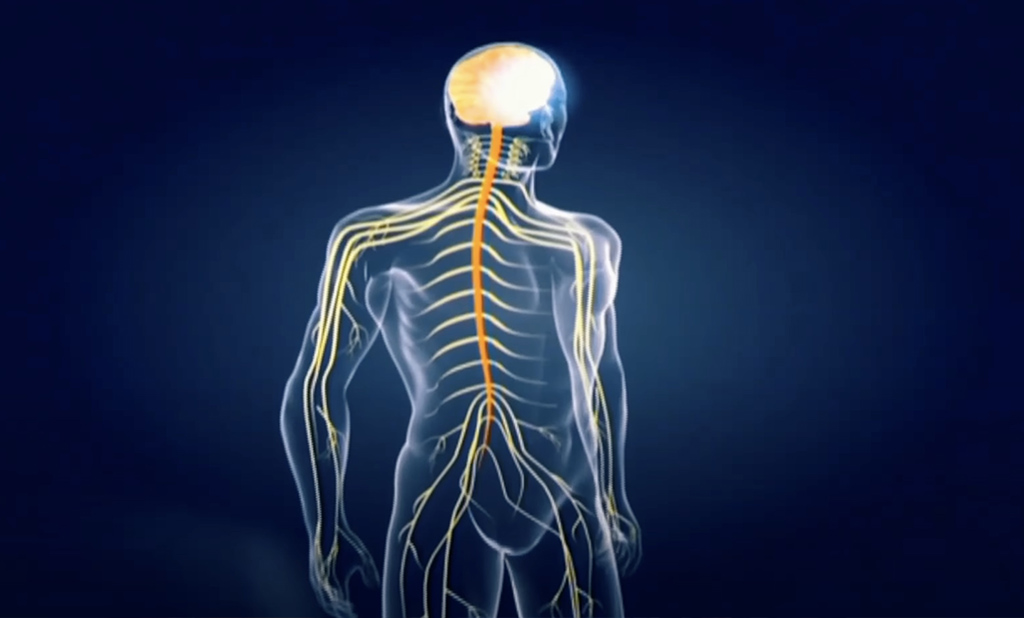
Ce sont des maladies génétiques qui détruisent la myéline (gaine des nerfs) du système nerveux. Lorsque cette gaine est abîmée, le courant ne passe plus et les messages nerveux sont interrompus, ce qui entraîne rapidement la dégradation de toutes les fonctions (vue, ouïe, motricité, mémoire…) et conduit très souvent au décès.
Les maladies :
• ALD/AMN
• Leucodystrophies indéterminées
• Maladie de Canavan
• Maladie de Krabbe
• Maladie de Refsum
• Maladie d’Alexander
• Maladie MLC
• MLD
• PMD et autres leucodystrophies hypomyélinisantes
• Syndrome Aicardi-Goutières
• Syndrome CACH
• LTBL
Quelques chiffres clés
• 3 à 6 enfants par semaine naissent atteints de leucodystrophie en France (20 à 40 en Europe).
• 48,8 millions d’euros investis au travers de 571 programmes de recherche sur les leucodystrophies.
• 15,7 millions d’euros consacrés à l’accompagnement des familles.